In the past decades, high-throughput
screening (HTS) applications have
been established in many fields of
drug development. Thus, suitable qualitative
and quantitative analytical methods
have become an important research
topic. Since more than 80% of drugs and
active ingredients are chiral substances,1
the development of fast and reliable analytical
methods for the determination of
the enantiomeric excess (ee%) is required
for improving drug efficacy and reducing
drug development costs. Common analytical
methods for the ee% determination
of chiral compounds include HPLC, GC,
and capillary electrophoresis (CE). Due
to their long analysis times and the use of
chiral columns and materials, these analytical
techniques cannot be used in high-throughput
experimentation.
Traverse et al.2 reviewed several techniques
for the determination of ee% in
HTS, including mass spectrometry. Lindner
et al.3 gave an overview on the use of
MS for ee% determination. A multitude of
different methods, such as host–guest complex
formation,4,5 ion–molecule reactions,6
collision-induced decay of diastereomeric
complexes,7 and parallel kinetic resolution
with mass-tagged auxiliaries, have
been established.8–13 In order to meet the
requirements of high-throughput systems,
it is necessary to combine sample preparation,
analysis, and data evaluation in a versatile
and easily adaptive processing system
with a minimum of manual work. In this
study, various substrates and auxiliaries
from different compound classes were
investigated and used in HTS systems.
Parallel kinetic resolution for ee% determination
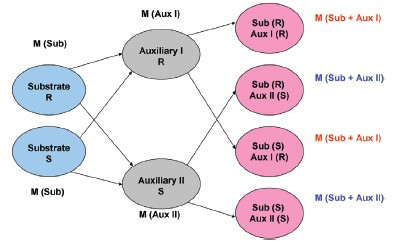
Figure 1 - General principle of parallel kinetic resolution.
Reetz et al.8 and Finn et al.9,10 described a
method for the ee% determination of chiral
catalysts using parallel kinetic resolution
and mass spectrometry. A chiral substrate
was derivatized with two pseudoenantiomeric
mass-tagged auxiliaries. Due to the
mass differences of the auxiliaries used, usually four products with two characteristic
masses are formed in this derivatization
process (see Figure 1 for the general
principle of parallel kinetic resolution).
Because of the different reaction kinetics
of the enantiomers and auxiliaries,
various quantities of the products
result. The ratios of the integrated peak
areas of both masses are characteristic
for each ee% value of the substrate.
Figure 2 shows an example with two
characteristic peaks for the determination
of (–)-carbobenzyloxy-L-proline
(100ee%). Finn et al.9,10 reported the
analysis of secondary alcohols and primary
and secondary amines as chiral substrates.
(S)-N-benzoyl-2-pyrrolidinecarboxylic acid,
(R)-N-(p-toluoyl)-2-pyrrolidinecarboxylic
acid, and their correlative “switched”
versions were used as mass-tagged pseudoenantiomeric
auxiliaries. In this study, the
method was extended and optimized for
further compound classes.
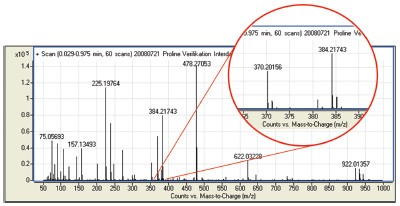
Figure 2 - Mass spectrum of derivated (–)-carbobenzyloxy-L-proline (100ee%).
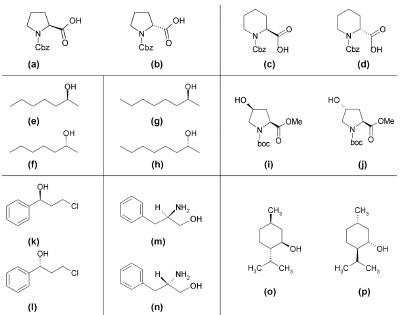
Figure 3 - Investigated chiral substrates: a) (–)-carbobenzyloxy-L-proline, b) (+)-carbobenzyloxy-D-proline, c) (S)-(–)-1-carbobenzyloxy-2-piperidinecarboxylic acid, d) (R)-(+)-1-carbobenzyloxy-2- piperidinecarboxylic acid, e) (S)-(+)-2-heptanol, f) (R)-(–)-2-heptanol, g) (S)-(+)-2-octanol, h) (R)-(−)-2-octanol, i) N-boc-cis-4-hydroxy-L-proline methyl ester, j) N-boc-trans-4-hydroxy-L-proline methyl ester, k)
(S)-(–)-3-chloro-1-phenyl-1-propanol, l) (R)-(+)-3-chloro-1-phenyl-1-propanol, m) (R)-(+)-2-amino-3-phenyl-1-propanol, n) (S)-(–)-2-amino-3-phenyl-1-propanol, o) (1R,2S,5R)-(–)-menthol, p) (1S,2R,5S)-(+)-menthol.
Experimental
Chemicals and reagents
Different carboxylic acids, alcohols, and
amino alcohols were analyzed (Figure
3). (–)-Carbobenzyloxy-L-proline (a),
(+)-carbobenzyloxy-D-proline (b), (S)-(–)-1-carbobenzyloxy-2-piperidinecarboxylic
acid (c), (R)-(+)-1-carbobenzyloxy-2-piperidinecarboxylic
acid (d), (S)-(+)-2-heptanol (e), (R)-(–)-2-octanol
(h), (1R,2S,5R)-(–)-menthol (o),
(1S,2R,5S)-(+)-menthol (p), (S)-(–)-3-chloro-1-phenyl-1-propanol (k), (R)-(+)-3-chloro-1-phenyl-1-propanol (l), N-boccis-4-hydroxy-L-proline methyl ester (i),
N-boc-trans-4-hydroxy-L-proline methyl
ester (j), and N,N′-dicyclohexylcarbodiimide
(DCC) were purchased from Sigma Aldrich
(Steinheim, Germany). (R)-(–)-2-heptanol
(f), (S)-(+)-2-octanol (g), (R)-(+)-2- amino-3-phenyl-1-propanol (m), (S)-(–)-2-amino-3-phenyl-1-propanol (n), and
4-(dimethylamino)-pyridine (DMAP) were
obtained from Fluka (Buchs, Switzerland);
methanol (HPLC gradient grade) was from
Roth (Karlsruhe, Germany), and dichloromethane
(HPLC grade) and toluene were
from AppliChem (Darmstadt, Germany).
Sample preparation
All tested substrates, with the exception
of (R)-(+)-2-amino-3-phenyl-1-propanol
and its corresponding enantiomer, were
solved in dichloromethane with a concentration
of 20 μmol/L. To create the calibration
curve, five stock solutions were prepared
with 100, 50, 0, –50, and –100ee%.
For the analysis of the carboxylic acids, an
auxiliary solution with (S)-(+)-2-heptanol
and (R)-(−)-2-octanol, each in a concentration
of 100 μmol/L, was prepared with
toluene. For the analysis of alcohols and
amino alcohols, the auxiliary solution contains
(–)-carbobenzyloxy-L-proline as well
as (R)-(+)-1-carbobenzyloxy-2-piperidinecarboxylic
acid in the same concentration.
Finally, a toluene solution, which
contains the reagents DCC (200 μmol/L)
and DMAP (2 μmol/L), was prepared.
(R)-(+)-2-amino-3-phenyl-1-propanol
and its corresponding enantiomer were
solved with a concentration of 0.2 μmol/mL in toluene. The corresponding auxiliary
solution contains (–)-carbobenzyloxy-L-proline and (R)-(+)-1-carbobenzyloxy-2-piperidinecarboxylic acid, each in a
concentration of 2 μmol/mL. The reagent
solution contains 5 μmol/mL DCC and 0.5
μmol/mL DMAP.
The derivatization was performed in 1-mL
vials from Agilent (Waldbronn, Germany)
and in 96-well multiple-well plates with
a well volume of 500 μL from Greiner-BioOne (Essen, Germany); 50 μL of the
solution with chiral substrates, 100 μL of the auxiliary solution, and 50 μL of the
solution with the reagents were added and
mixed in a thermo shaker (Thermomixer
comfort, Eppendorf, Hamburg, Germany)
for 1 hr at 20 °C followed by the addition
of 300 μL methanol. For the MS analysis,
the samples were diluted with methanol
(1:100, v/v).
Instrumentation
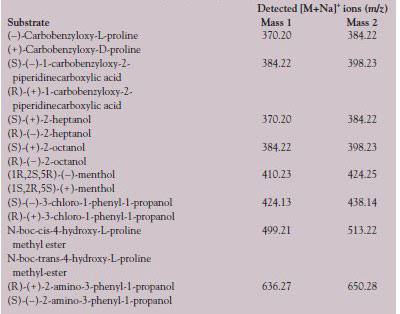
Table 1 - Characteristic masses (m/z) of the [M+Na]+ ions of the derivative pairs
The mass spectrometric analyses were
carried out on an Agilent LC-MS system consisting of a G1379B vacuum degasser,
G1312B binary pump, G1367C high-
performance
automated liquid sampler,
and G1969A time-of-flight mass spectrometer
(TOF-MS) with an electrospray ion
source. Five microliters of the sample solution
were injected with a prior needle wash.
The mobile phase had a flow of 1 mL/min
and consisted of methanol and water with
0.1% formic acid (90:10, v/v). The TOF-MS
was operated in positive ion mode with
the following parameters: nitrogen as nebulizer
and drying gas, nebulizer pressure of 35
psig, drying gas flow of 10 L/min, drying gas
temperature of 350 °C, capillary voltage of
4000 V, fragmentor voltage of 215 V, skimmer
voltage of 60 V, and octopol voltage
set at 250 V. The measurement was operated
using the SCAN mode in the range
of 50–1000 m/z. Data acquisition was done
using Mass Hunter Data Acquisition software
from Agilent. Table 1 shows the masses
(m/z) of the pairs of derivatives analyzed as
protonated ions [M+Na]+.