Hormones, such as androgens (AS),
estrogens (ES), and progestogens
(PS), play an important role in
organisms. AS, the main male sex steroids,
which are the critical factors responsible
for the development of the male phenotype
during embryogenesis and for the achievement
of sexual maturation at puberty, are
generally used as therapeutic agents for restoration
of the size and strength of muscle.1 It is known that estrogens have direct
effects on the areas of the brain that control
mood and cognition.2 Today, most formulations
of contraceptive pills contain ethinyl
estradiol, often in combination with a PS as
the active ingredient. Estrogens combined
with progestogens are also used to treat climacteric
syndrome.3
Drug abuse has an adverse impact on
aquatic products. Hormones can apply
to animal sex differentiation, shortening
the animal's growth cycle.4-6 Bisphenol
A caused apoptotic cell death in
swordtail testes and increased the mortality
of medaka eggs when males were
exposed to waterborne bisphenol A
before spawning.7 Diethylstilbestrol has
a range of actions on disruption of the
thyroid system in the sea bream, such
as encompassing the pituitary, thyroid
tissue, and deiodinases.8 Certain kinds
of hormones are often added to feed
to enhance the efficiency of aquatic
product breeding. Hormones are stable
and less susceptible to degradation,
yet still have strong biological activity
and potential carcinogenicity when
introduced into body tissues through
the food chain, inducing neutral fat,
immunodeficiency,
and osteoporosis.9,10
Many methods have been used to effectively
monitor and detect hormones
in animal tissues. These can generally
be divided into two major groups:
1) immunological methods , which
are highly selective due to the antibody–antigen specificity interaction,
although their applications in the samples
are limited to some extent due to
the instability of natural antibodies,
and can easily result in false positive
results,11,12 and 2) chromatographic
methods, such as HPLC,13,14LC-MS,15,16 and GC-MS,17,18 which
are the techniques most commonly
used for detecting hormones.
They, too, have some shortcomings
despite their commonality. HPLC
can only be used as a conventional
method, but not for characterization. GC-MS often requires a
derivatization step prior to chromatographic
separation, which is
mandatory for most of these compounds
because they are thermally
unstable, nonvolatile, or polar.
However, derivatization makes sample
preparation laborious and time-consuming,
and may lead to losses
or degradation of the target steroids.
UltraPerformance LC® (UPLC®) (Waters Corp., Milford, MA) with electrospray ionization-tandem mass spectrometry (UPLC-ESI-MS-MS)
has the virtue of efficient separation
and accurate characterization,
and has become the primary method
of determining hormones.19-24 The
authors established a method to
simultaneously determine 24 hormones
in aquatic products. The
result was satisfactory after systematized
optimization of sample pretreatment
methods and instrumental
analysis conditions.
Experimental
Materials
Cyproterone acetate and levonorgestrel
were purchased from Sigma Chemical
Co. (St. Louis, MO). 4-n-Octylphenol
was obtained from Supelco (Bellefonte,
PA). Bisphenol A, 4–nonylphenol,
diethylstilbestrol, estrone, 17α-ethinyl estradiol, 17β-estradiol, estriol, megestrol
acetate, trenbolone, norethisterone
acetate, ethisterone, progesterone,
zeranol, dihydrodiethylstilbestrol,
chlormadinone acetate, testosterone,
boldenone, 19-nortestosterone, methyltestosterone, dydrogesterone, and
dienestrol were all obtained from Dr.
Ehrenstorfer (Augsburg, Germany).
Methanol, methyl tertiary butyl ether,
and acetonitrile were all of HPLC grade
and from Tedia (Fairfield, OH). Water
was deionized ultrapure water. All other
reagents used in the experiment were of
analytical grade.
Preparation of stock and standard solutions
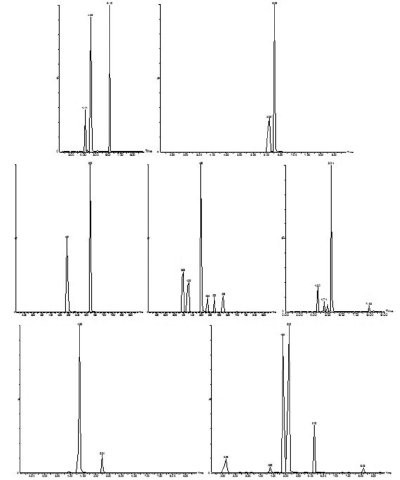
Figure 1 - Representative total ion chromatograms of 24 hormones. Names and retention times are as follows: estriol (2.63 min), trenbolone (3.98 min), boldenone (4.14 min), nandrolone (4.29 min), bisphenol A (4.15 min), zeranol (4.36 min), 17β-estradiol (4.38 min), testosterone (4.58 min),17α-Ethinyl estradiol (4.74 min), dihydrodiethylstilbestrol (4.88 min), methyltestosterone (4.91 min),ethisterone (4.98 min), diethylstilbestrol (5.13 min), dienestrol (5.24 min), estrone (5.25 min), levonorgestrel (5.36 min), dydrogesterone (5.73 min), cyproterone acetate (6.09 min), megestrol acetate (6.15 min), progesterone (6.22 min), norethisterone acetate (6.23 min), chlormadinone acetate (6.28 min), 4–nonylphenol (7.93 min), and 4-n-octylphenol (8.08 min).
Individual stock solutions of 100 µg/mL for all compounds were prepared in HPLC-grade methanol and kept at –20 °C. Standard solutions containing
all compounds were mixed and diluted
with methanol, and working solutions
of all compounds and calibration concentrations
were prepared by appropriate
dilution of the stock solutions on
the day of analysis. The total ion chromatograms
of 24 hormones (100 µg/L)
are shown in Figure 1.
Preparation and purification of samples
Samples (5 g) were homogenized for 30
sec in 20 mL of ethyl acetate, to which
3 mL 10% sodium carbonate was added,
and this was ultrasonicated for 10 min
at room temperature. The homogenates
were then centrifuged at 7000 rpm for
10 min. The supernatant was evaporated
at 40 °C and the residue was dissolved
in 10 mL methanol/water (1:9,
v/v), ready for loading.
The solid-phase extraction (SPE) process
with a Waters HLB column can be
summarized as follows: 1) Activation
with 5 mL ethyl acetate, 5 mL methanol (both steps at 3 mL·min-1); 2) loading 10-mL sample at 1.2 mL·min-1; 3) rinsing with 5 mL methanol/water (2:8,
v/v) and drying under vacuum for 5
min; and 4) elution with 10 mL methanol.
The eluent was then evaporated to
dryness under a gentle stream of nitrogen
at 40 °C and the residue was redissolved
with a volume of 1 mL of acetonitrile/ultrapure water (1:1, v/v) and
syringe filtered using a 0.22-µm filter
into an autosampler vial.
The UPLC-MS-MS system comprises
a Waters ACQUITY® UPLC system connected in-line to a Quattro Premier tandem mass spectrometer
(Waters).The column used in the
experiment was an ACQUITY UPLC
BEH C18 (2.1—100 mm, 1.7 µm particle
size) maintained at 40 °C. The
ACQUITY phase was A (acetonitrile)
and B (water). After sample injection
(10 µL), a linear gradient was programmed
for 8 min from 20:80 A–B
to obtain 95:5 A–B composition; then
the composition was held for 2 min.
Finally, the concentration of A was
decreased to 20% and held for 2 min.
The total analysis time was 12 min,
with 2 min required to reestablish and
equilibrate the initial conditions. The
flow rate was set at 0.25 mL·min-1 during
the chromatographic process, and
the temperature of the analytical column
was 40 °C. The entire eluent was
electrosprayed, ionized, and monitored
by MS-MS detection in multiple reaction
monitoring (MRM) mode. Ionization
was positive for AS and PS and
negative for ES. For this purpose, the
MS polarity was switched by time segments
according to the retention of
the target analytes. The flow rate and
temperature of the drying gas (N2)
were L·hr-1 and 350 °C, respectively.
The collision gas (Ar) flow was
0.12 mL/min, and the capillary voltage
was 2850 V. The dwell time was set at
100 msec.
Validation procedures
Standard calibration curves and QC
samples were analyzed over a period
of three consecutive days. Linearity of
calibration curves based on the analyte
area as a function of the nominal concentration
was assessed by weighted (1/C2) least-squares regression. Linearity
correlation coefficients were calculated
in the experiment.
Results and discussion
Optimization of LC–MS-MS
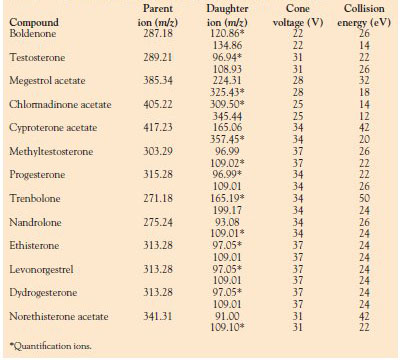
Table 1 - UPLC-ESI-MS-MS parameters for 13 hormones in ESI+ mode
Each tuning solution was introduced
into the electrospray source by direct
infusion (10 µL·min-1). The main ions
produced in MS and MS-MS were
identified in positive and negative
ionization modes. The diagnostic fragment
ions were selected and all mass
spectrometer parameters were optimized
to increase sensitivity. Tables 1
and 2 show the precursor and daughter ions for each steroid as well as the
optimum values of MS-MS parameters:
voltage of the first quadrupole for isolation
of the precursor ion and collision
energy for efficient fragmentation.
In the authors' study, two daughter
ions were routinely monitored. This
fulfills the recommendations of the
European Union (EU) concerning
identification, since two MRM transitions
from the ionized molecule of
the target compound give four points
in the scale—a value regarded as sufficient
for unequivocal identification.
The commonly used mobile-phase
compositions such as acetonitrile/water and methanol/water were optimized
in the experiment. In terms of
consumption and ion response, methanol
is a better choice.