The instability of small drug
molecules in biological fluids causes
serious bioanalytical challenges,
regardless of what type of powerful
analytical technology (i.e., HPLC-API [atmospheric pressure ionization]/ MS-MS) is employed for the
bioanalytical assay. As described in
the FDA guidance for industry,1 stability
is one of the basic parameters that
must be measured as part of a bioanalytical
method validation. It is recommended
that the stability of drug components
in spiked samples be
determined when establishing a bioanalytical
method.
In the early drug discovery stage,
extensive studies of drug stability are
not usually performed due to time
constraints. On the other hand, drug
hits that undergo rapid degradation
in plasma ex vivo may yield inaccurate
pharmacokinetic properties; thus
it is important to measure plasma stability
in the discovery setting. In the
drug development stage, in which
stability evaluation is obligatory,
instability of drug candidates in biological
samples seriously complicates
assay validation. This article provides
a brief review of the general methodologies
commonly utilized to stabilize
drug molecules that are unstable in
biological matrices.
Stability measurement
Traditional procedures for drug stability
measurement in spiked specimens
involve sample collection over a certain
period of incubation times followed by
sample preparation steps such as protein
precipitation to terminate the reaction
prior to instrumental determination. Previously,
a simple, semiautomated procedure
for screening drug stability in plasma
was developed in the authors’ laboratory
(Drug Metabolism and Pharmacokinetics
Dept., Schering-Plough Research
Institute, Kenilworth, NJ).2–4
The new procedure utilizes the
direct plasma injection method
based on a mixed-function column high-performance liquid chromatograph combined with tandem mass spectrometry (MS-MS).
5–9 Spiked plasma samples
containing drug compounds
were directly and sequentially
injected into a mixed-function
column for the on-line removal
of proteins and other macromolecules.
The drug components,
including their potential
degradation products, were then
eluted through chromatographic separation and monitored via
the tandem mass spectrometer
in one analytical procedure.
Drug stability in plasma indicated
by the change of mass
chromatographic peak area for
the test compounds was
observed to be a function of animal
species, time, and temperature.
The analytical results of the
drug stability experiments
obtained by the semiautomated direct
plasma injection method were found to
correlate with those obtained by the traditional
manual method using the protein
precipitation procedure. This
higher-throughput procedure also allows
plasma stability structure relationships
(PSSR) to be performed as part of
lead optimization.
Strategies for stabilization
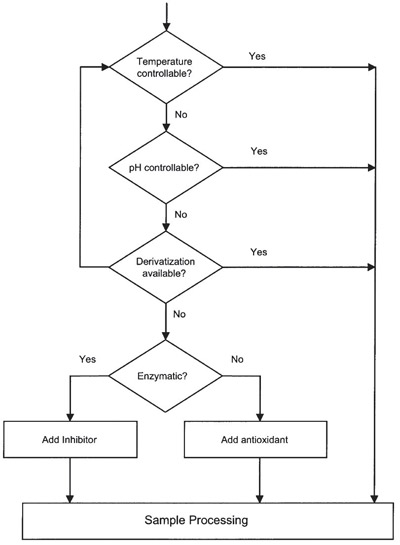
Figure 1- Flow chart of stabilization of small
molecules in biological fluids.
Instability of drug molecules in biological
samples is more common than
that in stock solution or extract after
sample processing. Biological samples
are much more complex in composition
than stock solutions. Thus, drug
molecules that readily degrade in biological
samples may still be stable in
stock solution. A major cause of analyte
instability in biological samples is
due to enzyme activity. In plasma, the
most prominent enzymatic activities
are the esterase activity for compounds
containing an ester group, and the
deamidase activity for compounds
containing an amide group.10 Esterases
are a heterogeneous family of enzymes
that catalyze the hydrolysis of esters
and amides to their corresponding carboxylic
acids. Carboxylesterases and,
to a lesser extent, cholinesterases are
important members of the esterase
family responsible for the metabolism
of drug compounds. As a result,
although many ester or amide bonds
are stable in solution at neutral pH,
they may be susceptible to hydrolysis
in plasma. The highest esterase activity
is usually in the liver, but it can also
be found in other tissues such as kidney,
brain, red blood cells, and plasma.
Drug stability in plasma may vary from
different animal species, depending on
the distribution of esterases in the
body. Therefore, drug stability is one of
the major concerns for the interpretation
of drug concentrations in biological
fluids. A general strategy for the
stabilization of pharmaceuticals in biological
samples is outlined in Figure 1.
Temperature control
The most universal approach to stabilizing
drug components is to lower the
temperature. Reduction of the temperature
normally slows down not
only enzymatic but also spontaneous
reactions. For spontaneous degradation
reactions, the effectiveness of
temperature control depends on the
activation energy. With an activation
energy of 20 kcal/mol, the reaction
rate is expected to be 10 times slower
when the temperature is decreased
from 22 °C to 0 °C (Arrhenius equation).
As an example, anthracenedione,
an antitumor reagent, was
found to be unstable in plasma,
degrading in an apparent first-order
reaction with t1⁄2 about one day at room
temperature. When refrigerated at
4 °C, the t1⁄2 of anthracenedione was
prolonged to six days.11 For enzymatic
reactions, however, the effectiveness
of this approach depends on the magnitude
of reduction in enzyme activity
at lower temperature.
Temperature control also has implications
for long-term plasma storage in
the frozen state. Using NMR spectroscopy,
it was demonstrated that there
is a 20-fold decrease in the amount of
unfrozen water in frozen human plasma
when the temperature is lowered from
–20 °C to –80 °C.11 As a consequence,
the epimerization kinetics of moxalactam
was observed to be reduced with
the smaller amount of unfrozen water.12
Cisplatin, another anticancer drug for
the treatment of solid tumors, and its
monohydrated complex are not stable
at –25 °C but are stable at –70 °C for at
least three weeks.13
pH adjustment
The second approach, pH control,
takes advantage of the fact that most
enzymes have a narrow range of working
pH. Albumin possessing weak
hydrolase activity in its IIIA subdomain
is the most abundant protein in
animal plasma. It binds to a number
of drug molecules with various affinities,
conferring stability to certain
compounds that are otherwise unstable
in plasma. Fura et al.14 reported
that the pH of ex vivo plasma, bile,
and urine changes during long-term
storage and sample preparation procedures
such as protein precipitation, centrifugation, ultrafiltration, and evaporation. The pH shifts in biological
species will significantly affect
protein-binding measurement and
alter the disappearance rate for pH-sensitive
compounds such as acylglucuronides.
15 The pH may have a
strong impact on both acid and base
catalyzed enzymatic and nonenzymatic
reaction.16 The addition of a
small amount of appropriate buffers
such as phosphate, citrate, and bicarbonate
into biological samples to
maintain an optimum pH was shown
to be an effective way to prevent
degradation.14,17 One complication
for pH adjustment is that at very
acidic pH, plasma proteins may precipitate,
causing problems for sample
storage and preparation.
Derivatization
One of the analytical purposes for the
derivatization of pharmaceuticals is to
ease the stability issue in biological samples.
For example, drug molecules containing
a sulfhydryl group (the thiol
compound) are generally not stable in
plasma. The thiol group is a strong
nucleophile that may react with cystine
residues in plasma protein or glutathione
to form disulfide bonds,
depending on the solution pH and oxidation
potential. Derivatization is one
of the useful approaches employed to
stabilize this class of compounds. In this
approach, the thiol compound reacts
with an alkylating agent to form
Michael addition derivatives.18–20 Both
methyl acrylate (MA) and Nethylmaleimide
have been used as the
derivatizing agent. Derivatization with
methyl acrylate has the advantage over
that with N-ethylmaleimide, which creates
a new chiral center to yield two
diastereometric derivatives. As an
example, Jemal and co-workers reported
work on the simultaneous determination
of omapatrilat and its four circulating
metabolites.19Omapatrilat has a free
thiol and therefore is extremely unstable
in human blood and plasma at room
temperature. Two of omapatrilat’s
metabolites also contain a thiol and
have similar plasma stability profiles.
The reaction between MA and omapatrilat
in plasma was conducted on ice to
reduce the loss of analytes during sample
processing. The resulting MA–omapatrilat
products in human plasma were
stable for at least 6 hr at room temperature
and for more than 24 hr at 4 °C.
The derivatives in plasma were then
extracted for analysis.