It was previously reported1 that when Direct Analysis in Real Time (DART™) (JEOL USA, Inc., Peabody, MA) mass spectrometry was applied to the analysis of very pure alcohols, their mass spectra were unexpectedly complicated. The plethora of peaks was found to be a DART-induced artifact that resulted from the open-air nature of the technique. This problem was solved by converting the alcohol into its corresponding carbamate via an N-hydro-C-alkoxy-addition reaction (reaction 1) with phenyl isocyanate:
ROH + C6H5NCO → RO−C(O)NHC6H5 (1)
Although this reaction conveniently allows alcohols to be DART’ed, the reaction time ranges from 1 to 16 hr. This obviously led the authors to test the hypothesis that shorter reaction times can be achieved by using activated phenyl isocyanates with proper ring substitution. It is now timely to report these results.
Experimental
Chemicals were either synthesized in-house or purchased from Aldrich Chemical Co. (St. Louis, MO). The alcohol precursors were converted into their corresponding carbamates using the N-hydro-C-alkoxy-addition reaction. The reaction occurs at room temperature without by-product formation and is simple to perform. Ten to fifty microliters of the alcohol was pipetted into a small vial and then weighed; 1000 μL of hexane was added, and then a 1.1 molar excess of substituted phenyl isocyanate was introduced. Because the O-alkyl carbamate product is not soluble in hexane, it is automatically purified as it crystallizes out of solution. An additional benefit of the reaction scheme is that the end point of the reaction is conveniently indicated by a vial full of crystals.
The experimental apparatus consisted of a DART ion source (model 100-001-JL1) and modified AccuTOF mass spectrometer (model JMS-100TLC) (JEOL USA, Inc.). Excited-state species and metastable ions and neutrals are the working reagent in the DART method.2 The use of excited-state species as a replacement for the radioactive source in chemical agent monitors was first conceived by Dr. Laramée in 2001. Working prototypes were developed by Dr. Robert B. Cody and Dr. Laramée, which were publicly disclosed on April 14, 2003 in two patent applications as the first ambient mass spectrometric method.3
Accurate exact mass measurements are important in order to assign correct and unambiguous elemental compositions to mass spectral peaks. The average mass accuracy was ±0.00004 Da by summing the mass deviations. By way of contrast, the instrument manufacturer claims a mass accuracy of ±0.002 Da but recently reported measured mass in disagreement with calculated mass values of approximately 0.007 Da, with even larger deviations observed at masses below 200 and above 600 Da.4 This instrument behavior has not been observed on any of the authors’ three DART/AccuTOF systems. The authors attribute this to methodical operation and setup, both of which appear to be essential for obtaining meaningful exact mass measurements on the AccuTOF mass spectrometer.
Results and discussion
If an organic chemist were given a choice of which items to be stranded with on a desert island, he or she would most certainly choose alcohols. From these, a host of other compounds could be made and used as solvents in which the reactions occur and from which products are recrystallized. Thus, alcohols are useful, but alcohols cannot be analyzed well by DART.
This problem was solved by using phenyl isocyanate to convert the alcohol into its corresponding carbamate. This conversion not only simplifies the mass spectrum, but also increases its analytical detection sensitivity 20-fold. The reaction is quantitative, as evidenced by the fact that the mass spectrum did not contain the alcohol starting material. For any reaction in equilibrium, removal of the final product drives the reaction to completion. This was achieved by careful selection of reaction solvents, in which the starting reagents are soluble but the final product is insoluble.
Although conversion of the alcohol into a carbamate by phenyl isocyanate solved the analysis problem, the somewhat long reaction times prompted the authors to explore activated phenyl isocyanates that had ring substitutions. The reaction times of neo-pentyl alcohol with 2-nitro-4-(trifluoromethyl) phenyl isocyanate, phenyl isocyanate, and N,N-dimethylamino phenyl isocyanate were compared. 2-Nitro-4-(trifluoromethyl) phenyl isocyanate reacted almost instantaneously with product crystals forming as soon as the reagents were combined. Phenyl isocyanate required a few hours to react, and N,N-dimethylamino phenyl isocyanate took even longer.
It was also discovered that irradiating the reaction vial with light (hν) increased the reaction rate by two orders of magnitude. A standard high-intensity tungsten–halogen lamp was used. No attempt was made to determine the wavelength responsible for the catalytic effect. Nevertheless, the result is useful because light is an ideal catalyst since it does not leave a catalytic residue that must be removed prior to analysis. The effect of warming the reaction vial to 80 °C in a hot sand bath was also explored and was found not to be as effective as the light irradiation.

Figure 1 - Mass spectrum of neo-pentyl-N,N-dimethylamino phenyl carbamate. Note the low relative intensity of the mass spectrum (77K counts full scale) despite the presence of two nitrogen atoms with large proton affinities. The protonated neutral (MH+) is the base peak, but a molecular radical cation (M+•) and a series of oxygen adducts are also seen in abundance. The peak labeled “NCO” at mass 163 is unreacted isocyanate.
Neo-pentyl-N,N-dimethylamino phenyl carbamate gave the expected MH+ molecular cation, but also produced a series of oxygenated adducts and an interfering M+• molecular radical cation. A rather anemic mass spectral abundance was also seen (Figure 1). This was an unexpected result because tertiary amines typically have excellent ionization efficiencies by atmospheric ionization techniques.
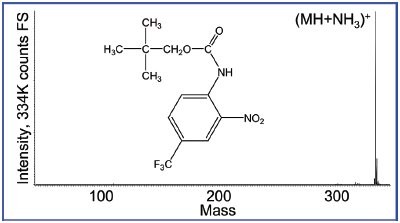
Figure 2 - Mass spectrum of neo-pentyl-2-nitro-4-(trifluoromethyl) phenyl carbamate. Note that the base peak is now more than four times its intensity compared to Figure 1.

Figure 3 - Nucleophilic attack by the alcohol onto the carbonyl carbon of phenyl isocyanate. The two perpendicular π-electron systems in the isocyanate moiety are responsible for the unusual charge distribution indicated and account for the ion formation difference seen in Figures 1 and 2.
On the other hand, neo-pentyl-2-nitro-4-(trifluoromethyl) phenyl carbamate gave a single (MH+NH3)+ peak of high relative intensity (Figure 2). Acylation of alcohols with isocyanates by N-hydro-C-alkoxy-addition (reaction 1) is poorly understood,5 although the oxygen of the alcohol is unquestionably attacking the carbonyl atom of the isocyanate (Figure 3). Virtually all of the known reactions like reaction 1 are heterolytic processes. In such circumstances, electron withdrawal by substituents on the phenyl ring will magnify the positive charge on the carbonyl carbon atom (Figure 3), thus enhancing nucleophilic attack and consequently speeding up the reaction rate. This hypothesis was tested by reacting the same alcohol with various ring-substituted phenyl isocyanates. Pinacolyl alcohol was chosen for study because of its use in the authors’ synthesis of O-pinacolyl methylphosphonofluoridate (Nerve Agent GD).

Figure 4 - DART spectra of the phenyl substituted carbamates. Hammett substituent constants (σ) indicate increasing electron withdrawing ability: a) Pinacolyl-3-nitrophenyl carbamate, σ = 0.71; b) pinacolyl-2-nitro-4-(trifluoromethyl) phenyl carbamate, σ = 0.69; c) pinacolyl-4-nitro-2-(trifluoromethyl) phenyl carbamate, σ = 0.67; d) pinacolyl-4-(trifluoromethyl) phenyl carbamate, σ = 0.53; e) pinacolyl-3,4-difluorophenyl carbamate, σ = 0.25; f) pinacolyl phenyl carbamate, σ = 0.0; g) pinacolyl-3-methoxyphenyl carbamate, σ = 0.12; h) pinacolyl-4-methoxyphenyl phenyl carbamate, σ = –0.28; i) pinacolyl-N,N-dimethylaminophenyl carbamate, σ = –0.63. Note the increasing spectral complexity as the Hammett substituent constant becomes progressively more negative in value.
Nine pinacolyl phenyl-substituted carbamates were synthesized and their DART spectrum measured (Figure 4). The spectra were arranged according to the free energy electron withdrawing ability of the substituted-phenyl ring using Hammett substituent constants.6 The carbamates with the greatest electron withdrawing substituents were pinacolyl-3-nitrophenyl carbamate, pinacolyl-2-nitro-4-(trifluoromethyl) phenyl carbamate, and pinacolyl-4-nitro-2-(trifluoromethyl) phenyl carbamate, having Hammett substituent constants of 0.71, 0.69, and 0.67, respectively. These spectra are dominated by intense (MH+NH3)+ molecular cation peaks (Figure 4a–c). Substituents of intermediate electron withdrawing ability begin to produce MH+ molecular cations in addition to intense (MH+NH3)+ molecular cations (Figure 4d–f). But when the Hammett substituent constant approaches 0.12 or less, the molecular radical cation, M+•, becomes increasingly dominant and the spectra become more and more complicated (Figure 4g–i). This effect is seen for pinacolyl-3-methoxyphenyl carbamate, pinacolyl-4-methoxyphenyl phenyl carbamate, and pinacolyl-N,N-dimethylamino phenyl carbamate.
Conclusion
Conversion of an alcohol to its carbamate not only simplifies the mass spectrum but also increases its analytical detection sensitivity. The best reagents for the authors’ new reaction method are 2-nitro-4-(trifluoromethyl) phenyl isocyanate, 4-nitro-2-(trifluoromethyl) phenyl isocyanate, and 3-nitrophenyl isocyanate. This conversion reaction is fast and quantitative.
References
- Laramée, J.A.; Durst, H.D.; Nilles, J.M.; Connell, T.R. Alcohols can now be analyzed by a direct analysis in real-time method: applications for chemical warfare agent synthesis. Am. Lab. 2009, 40(4), 24–7.
- Laramée, J.A.; Durst, H.D.; Connell, T.R.; Nilles, J.M. Detection of chemical warfare agents on surfaces relevant to homeland security by direct analysis in real-time spectrometry. Am. Lab. 2008, 40(8), 16–20.
- Laramée, J.A.; Cody, R.B.; Nilles, J.M.; Durst, H.D. Forensic Analysis on the Cutting Edge. In Forensic Application of DART; Blackledge, R.D., Ed.; Wiley-Interscience: Hoboken, NJ, 2007; pp 175–95.
- Kpegba, K.; Spadaro, T.; Cody, R.B.; Nesnas, N.; Olson, J.A. Analysis of self-assembled monolayers on gold surfaces using direct analysis in real time mass spectrometry. Anal. Chem. Corr. 2007, 79, 5479–83.
- March, J., Ed. Advanced Organic Chemistry, 4th ed.; Wiley-Interscience: New York, NY, 1992, inter alia.
- Lowry, T.H., Richardson, K.S. Mechanism and Theory in Organic Chemistry; Harper and Row: New York, NY, 1976; pp 60–71 and 337–99.
Dr. Laramée is Principal Scientist, EAI Corp., now a subsidiary of Scientific Applications International Corp., P.O. Box 68, Aberdeen Proving Ground, Edgewood, MD 21010, U.S.A.; tel.: 410-436-2987; fax: 410-436-7317; e-mail: [email protected]. Dr. Durst is Research Chemist and Head, Chemical Methodology Team, U.S. Army Edgewood Chemical Biological Center, Aberdeen Proving Ground, Edgewood, MD. Dr. Nilles and Ms. Connell are Senior Scientists, Science & Engineering Technologies, Inc., Springfield, VA, U.S.A. The authors gratefully acknowledge the facility support of the Research & Technology Directorate at the Edgewood Chemical Biological Center (ECBC), and fiscal support from the Agent Fate Program (DTO.42), BA07RAS101 by the Joint Science and Technology Office (JSTO) in the Defense Threat Reduction Agency (DTRA), and the DoD Chemical and Biological Defense Program. This document is UNCLASSIFIED and cleared for public release through American Laboratory.