Improving protein solution behavior is critical in the field of structural biology since it impacts protein crystallization and the ability to determine protein structures by solution nuclear magnetic resonance (NMR) methods. Many proteins remain intractable to these powerful techniques because they are inherently unstable or have a tendency to form aggregates under typical buffer conditions. Dynamic light scattering (DLS) is a powerful tool capable of monitoring the aggregation state of protein molecules in solution, while also assisting in the initial generation and further optimization of protein crystals, through efficient and reliable optimization of protein solution behavior.1,2 More monodisperse protein samples often result in more crystallization hits in screening, and sometimes result in crystals with better diffracting properties compared to crystals arising from polydisperse protein samples.3 DLS is particularly effective if applied in conjunction with other methods used for characterizing protein behavior, including native polyacrylamide gel electrophoresis (PAGE) and sizeexclusion chromatography (SEC). The National Research Council-Biotechnology Research Institute (NRC-BRI) implements DLS and SEC to improve protein solution behavior for crystallization.
Background
NRC-BRI is the largest Canadian research facility devoted to biotechnology. Located in Montréal, Canada, the institute promotes, assists, and conducts leading-edge R&D in the fields of health, bioprocessing, and the environment. In the field of life sciences, NRC-BRI’s areas of expertise include drug discovery, bioprocess development, and scaleup. Environmental biotechnology involves the development of biologically based processes for protecting, cleaning, and monitoring soil, water, and air.
NRC-BRI scientists and engineers carry out research activities in five major research areas: the Health Sector invests considerable effort in cancer therapies and infectious diseases; the Bioprocess Sector uses the latest technologies and equipment for bioprocess development, and the Environment Sector addresses the twin challenges of environmental protection and clean manufacturing.
In addition to the diverse specialized services it provides, NRC-BRI also maintains active partnerships with the industrial and university sectors and with government agencies. These partnerships are based on formal collaborative agreements, research and service contracts, or technology transfer agreements. In addition, NRC-BRI houses an Industrial Partnership Facility (IPF) that accommodates biotechnology companies requiring laboratory space for their R&D activities.
Using DLS and SEC to improve protein behavior for crystallization
NRC-BRI utilizes DLS screening of purified protein samples routinely in conjunction with protein crystallization experiments. While some purified samples reveal monodisperse behavior in a standard buffer, many samples do not, and require additional treatment. In some instances, samples are initially polydispersed by DLS and are further purified by SEC, usually using a Hi-Load Superdex-75 or Superdex-200 gel filtration column (GE Healthcare, Mississauga, Ontario, Canada) running on a Fast Protein Liquid Chromatography (FPLC) system. Following chromatographic analysis, the behavior of protein peaks eluting in the fractionation range for the particular column is analyzed using a DLS instrument run at ambient temperature. The SEC column can remove larger protein aggregates that contribute to protein polydispersity, thereby yielding a more homogeneous sample in solution. It has been the experience of NRC-BRI that some of these monodisperse protein samples go on to yield crystals following crystallization screening.
In their experiments, NRC-BRI scientists have used the Aktä Explorer FPLC system (GE Healthcare) for column chromatography, and the DynaPro plate reader (Wyatt Technology, Santa Barbara, CA) for DLS measurements.
Application example
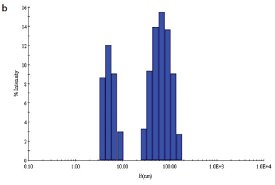
Figure 1 - b) DLS of C. jejuni double-mutant protein sample prior to SEC
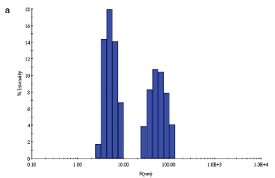
Figure 1 - a) DLS of C. jejuni single-mutant protein sample prior to SEC.
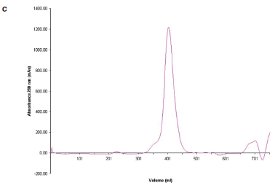
Figure 1- c) SEC chromatogram showing elution profile of C. jejuni single-mutant protein.
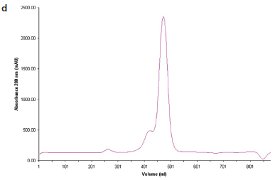
Figure 1 - d) SEC chromatogram showing elution profile of C. jejuni double-mutant protein.
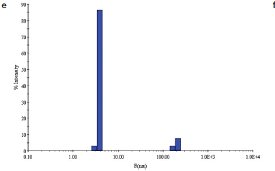
Figure 1 - e) DLS of C. jejuni single-mutant protein sample following SEC.
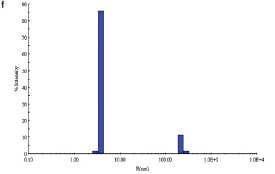
Figure 1 - f) DLS of C. jejuni double-mutant protein sample following SEC.
Two site-specific mutants of a biosynthetic protein from Campylobacter jejuni were prepared in order to improve the diffraction properties of these crystals. The initial DLS of these samples in buffer—10 mM (4-(2-hydroxyethyl)-1- piperazineethanesulfonic acid [HEPES]), pH 7.5, 0.5 M NaCl—is shown in Figure 1a and b. In order to remove protein aggregates, the samples were chromatographed on a Superdex-200 gel filtration column using a buffer consisting of 20 mM Tris-HCl pH 8.0, 200 mM NaCl, and 5 mM dithiothreitol (DTT) (Figure 1c and d). In both cases, a small void volume peak, as well as a shoulder on the leading side of the primary peak, can be observed. The major peak corresponds to the dimer form of the enzyme. SEC improved the solution behavior of these samples, as shown by their DLS behavior following chromatography (Figure 1e and f). Protein fractions from the main peak following SEC were pooled, concentrated to 5 mg/mL, and crystallized successfully. Recently, the structure of the double-mutant form of the enzyme was determined at 2.1-Å resolution (unpublished results).
Using additive screens to improve protein monodispersity
Table 1 - Additives used in optimization of protein behavior by DLS
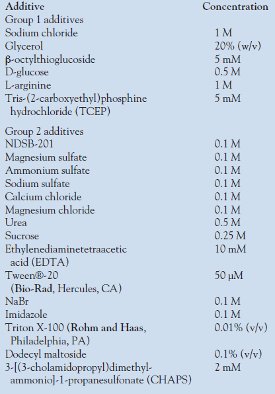
A second approach utilized by NRC-BRI scientists is to apply the concept of protein solubility screening to improve protein behavior, using DLS as a tool to monitor protein behavior. A number of solubility screens have been developed to assess and improve protein solubility, although in most cases this is determined by inspection of the precipitation pattern of the protein as viewed under a microscope4–7 or by gel electrophoresis.8,9
NRC-BRI scientists have developed a similar approach, but rather than examining precipitation patterns or electrophoretic behavior, they examine proteins using DLS. Especially useful in this regard is an instrument equipped with a plate reader, since it allows for the examination of a number of different additives in a relatively short time span.
In this type of experiment, an aliquot (50 μL) of protein (3–6 mg/mL) in the initial buffer (20 mM Tris pH 7.5, 50 mM NaCl, and 5 mM DTT) is tested with a series of additives, and the DLS profiles are compared at the same protein concentration. These additives are divided into two groups (Table 1); those in the first group are primary additives, since they are more likely to have a positive influence on the behavior of the protein. Additives in the second group are secondary additives, which are examined if none of the additives of the first group shows a positive effect. These can also be combined with the additives of the first group in those cases in which more than one additive may act synergistically to improve solution behavior. Additives that have a positive influence on protein behavior, i.e., reduced polydispersity, are then utilized as additives, either in the original lysis buffer used to prepare the protein extract, or as additives to be added to the purified protein prior to crystallization screening.
Application examples
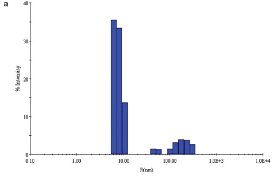
Figure 2 - a) DLS of E. coli YdiF protein in the absence of coenzyme A.
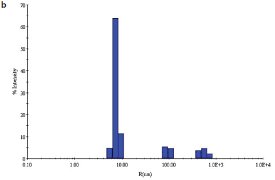
Figure 2 - b) DLS of E. coli YdiF protein in the presence of 1 mM coenzyme A.
NRC-BRI scientists have optimized the solution behavior of E. coli YdiF, a putative acyl-CoA (coenzyme A) transferase. The initial buffer contained 50 mM Tris pH 7.5, 0.2 M NaCl, 5% (w/v) glycerol, 5 mM DTT, and 50 mM imidazole with protein at 8 mg/mL. Tests with various additives showed that the addition of coenzyme A, a cofactor for this enzyme, at a concentration of 1 mM, decreased the apparent polydispersity of the sample from 23% (Figure 2a) to 13% (Figure 2b). While both samples of protein yielded crystals, albeit under different conditions, the scientists were ultimately able to trap the coenzyme A at the YdiF active site as a thioester intermediate by including various coenzyme A derivatives during crystallization.10 This structure represented the first crystal structure of a Family I coenzyme A transferase bound to coenzyme A.
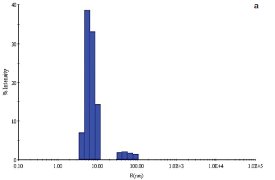
Figure 3 - a) DLS of E. coli YgjO protein in the absence of S-adenosylmethionine.
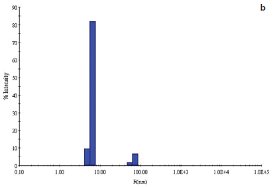
Figure 3 - b) DLS of E. coli YgjO protein in the presence of 1 mM S-adenosylmethionine.
A second example is with the E. coli protein YgjO, a putative 16S rRNA methyltransferase, expressed as an N-terminal glutathione S-transferase (GST)- fusion protein. Following cleavage of the GST, the sample was concentrated to 6 mg/mL in a buffer containing 20 mM Tris pH 7.5, 0.2 M NaCl, 4% (w/v) glycerol, and 5 mM DTT. The sample polydispersity was 26% (Figure 3a) and decreased to 9% (Figure 3b) upon addition of the substrate S-adenosylmethionine (SAM) at a concentration of 1 mM. Screening of the same protein sample in the presence of 1 mM SAM yielded a single crystallization hit.
Conclusion
Two different approaches are available to improve the behavior of purified proteins prior to crystallization trials with the aid of a DLS instrument. Highly purified, stable protein samples containing a minimum of aggregated species are often necessary to obtain well-diffracting crystals that are suitable for structural analysis. As with many aspects of protein chemistry, one or the other of the two approaches described in this article may be suitable depending on the particular protein at hand, and specific parameters have to be determined empirically. The second approach, namely the screening of additives to improve protein monodispersity, can become a routine application in laboratories only when the DLS instrument is equipped with a plate reader. The plate reader capable of working with 96- or 384-well plates offers the potential to rapidly examine a large number of samples using a minimum quantity of protein. It is also well suited for investigating individual fractions following chromatography, or with a variety of additives.
References
- Ferré-D’Amaré, A.; Burley, S.K. Use of dynamic light scattering to assess crystallizability of macromolecules and macromolecular assemblies. Structure1994, 2, 357–9.
- Wilson, W.W. Light scattering as a diagnostic for protein crystal growth—a practical approach. J. Struct. Biol.2003, 142, 56–65.
- Zulauf, M.; D’Arcy, A. Light scattering of proteins as a criterion for crystallization. J. Cryst. Growth1992, 122, 102–6.
- Collins, B.K.; Tomanicek, S.J.; Lyamicheva, N.; Kaiser, M.W.; Mueser, T.C. A preliminary solubility screen used to improve crystallization trials: crystallization and preliminary X-ray structure determination of Aeropyrum pernix flap endonuclease-1. Acta Cryst.2004, D60, 1674–8.
- Collins, B.; Stevens, R.C.; Page, R. Crystallization optimum solubility screening: using crystallization results to identify the optimal buffer for protein crystal formation. Acta Cryst.2005, F61, 1035–8.
- Izzac, A.; Schall, C.A.; Mueser, T.C. Assessment of a preliminary solubility screen to improve crystallization trials: uncoupling crystal condition searches. Acta Cryst.2006, D62, 833–42.
- Ducat, T.; Declerck, N.; Gostan, T.; Kochoyan, M.; Déméné, H. Rapid determination of protein solubility and stability conditions for NMR studies using incomplete factorial design. J. Biomol. NMR2006, 34, 137–51.
- Bondos, S.E.; Bicknell, A. Detection and prevention of protein–protein aggregation before, during and after purification. Anal. Biochem.2003, 316, 223–31.
- Lindwall, G.; Chau, M.-F.; Gardner, S.R.; Kohlstaedt, L.A. A sparse matrix approach to the solubilization of overexpressed proteins. Prot. Eng.2000, 13, 67–71.
- Rangarajan, E.S.; Li, Y.; Ajamian, E.; Iannuzzi, P.; Kernaghan, S.D.; Fraser, M.E.; Cygler, M.; Matte, A. Crystallographic trapping of the glutamyl-CoA thioester intermediate of Family I CoA transferases. J. Biol. Chem.2005, 280, 42,919–28.
The authors are with the National Research Council-Biotechnology Research Institute (NRC-BRI), 6100 Royalmount Ave., Montréal (Québec) H4P 2R2, Canada; tel.: 514-496-6321; fax: 514-496-5143; e-mail: [email protected].