The simultaneous detection of several peptides or proteins
in mixtures is an integral part of expression proteomics
investigations. Strategies that enable the
identification and characterization of several proteins
or peptides simultaneously from complex mixtures
with minimal recourse to sample cleanup or separation
stages prior to detection are ideally desired for
high-throughput proteomic investigations. The
advent of soft ionization mass spectrometric techniques,
such as matrix-assisted laser desorption ionization-mass spectrometery (MALDI-MS) and electrospray ionization-mass spectrometery (ESI-MS), has
enabled different strategies to be developed based on
these techniques.1–4 In particular, the higher mass
accuracy, sensitivity, and better reproducibility of
analysis possible with ESI- (and nanospray-) MS have
allowed its broader application in expression proteomic
investigations. It has been employed in both
“top-down” (involving analysis of “intact” proteins by tandem mass spectrometry without recourse to enzymatic
proteolytic digestions)5,6 and “bottom-up”
strategies (involving the detection of peptides from
digested proteins)7,8 for proteomic characterizations.
In ESI-MS analysis, several factors influence signal
detection, including the analyte concentration, pH
and ionic strength of the medium, nature of the analyte,
and instrumental parameters.9 For proteins,
these factors are known to influence the intensity
and charge state distribution of the protein peaks,
even when the proteins are analyzed in isolation.9–12
When considering the analysis of proteins or peptides
in mixtures, the ionization of the individual
components and the presence of other components
can affect the detection of any individual peptide or
protein.13 Although it has been recognized that several
factors contribute to the formation of gas-phase
ions in ESI-MS, understanding the behavior of ions
for the efficient analysis of proteins or peptides in
mixtures is far from complete. Once gas-phase ions
are generated in the source, they must still traverse
the source–analyzer interface before being detected.
This presents an additional set of factors that can
influence the protein or peptide signals detected.
Skimmer voltages, pressure, and ion transmission
optics have all been shown to be influential in the
detection of appropriate protein signals.10,14–17 The
tuning of instrumental settings in the source and
source–analyzer interface is thus likely to influence
the detection of protein signals in mixtures.
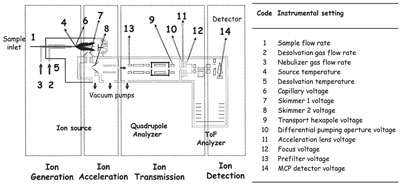
Figure 1 - Schematic showing instrumental configuration of the ESI-QToF mass spectrometer used in the study. The areas associated
with the 14 instrumental settings are numbered in the figure and listed in the table alongside. The regions of spectrometer associated with
ion generation, acceleration, transmittance, and detection are also indicated.
Investigations conducted by the author and colleagues18,19
have demonstrated this aspect with a
cocktail of five proteins in the mass range of 5–17
kDa using a Micromass (now Waters, Milford, MA)
ESI-QToF mass spectrometer. The mixture of five
proteins, including insulin (5733 Da), ubiquitin
(8565 Da), cytochrome c (12361 Da), lysozyme
(14309 Da), and myoglobin (16951 Da), was dissolved
in equimolar concentrations and analyzed in
acidic conditions in the positive ion mode. The concentration
used was low enough to maintain conditions
of excess charge and linearity of signal response,
so that any change in signal observed is not due to
charge limitations or concentration differences but
due solely to the nature of the protein and its behavior
under a given set of instrumental conditions.
Fourteen instrumental settings (Figure 1) that influence
ion generation, acceleration, transmission, and
detection within the mass spectrometer were studied.
Over 400 combinations of the instrumental settings
were analyzed.
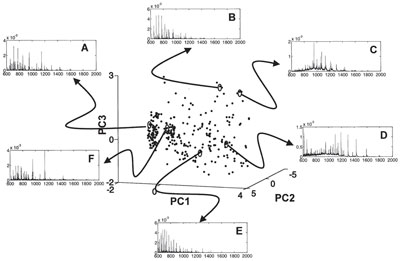
Figure 2 - Pseudo 3-D plot of the variable search space in the first three principal components (PC1,2,3) showing the distribution of
the trials where each trial indicates a set of instrumental conditions. Trials in which uniform detection of all five proteins (A) and the
preferential detection of cytochrome c (B), ubiquitin (C), lysozyme (D), myoglobin (E), and insulin (F) in the mixture are circled, and
their corresponding mass spectrum is shown. The difference in the spectra between the different cases can be clearly seen.
The responses of the individual proteins when analyzed
in isolation differed from their responses when analyzed in the mixture, even for a given instrumental
setting.18 The instrumental settings had a considerable
effect on the detection of the protein peaks, so
much so that under certain conditions selective
detection of the individual proteins in complete
exclusion of the others in the mixture was possible.19
The protein signals were influenced to varying
degrees under different instrumental settings. An
analysis of the instrumental settings in relation to
the responses shows that it is possible to identify
areas in the search space in which the conditions
permit selective detection of one or more proteins in
the mixture and in which all five proteins could be
evenly detected (Figure 2).
The problem of identifying unique values of the settings
for detecting the proteins preferentially or
evenly is complicated by its epistatic nature (i.e., the
effect of a given setting can depend on the values of
the others). However, it should be possible to optimize
the settings for a desired effect, as has been
demonstrated by the author and colleagues for the
case of uniform detection of all five proteins in the
mixture.18 Given the number of influencing factors,
their levels, and the resulting combinations to experiment,
heuristic methods will need to be employed,20
which seek solutions that approach an optimum but
cannot be guaranteed to find it, such as evolutionary
algorithms (EAs), in navigating the search spaces to
find regions of optimality for a desired effect.
The utility of such computational methods is
demonstrated by the fact that for the uniform detection
of all five proteins in the mixture, the use of an
EA in the search on a search space that was a nominal
1014 experiments resulted in convergence
toward optimality, even before 500 of them were
evaluated.18 The added advantage of such computational
strategies is that in addition to being
exploratory, they can be explanatory in that they
can sometimes provide an explanation for the optimal
behavior. In this particular experiment, it was
noted that much of the desired result of the uniform
detection of all five proteins arose from maintaining
a defined skimmer cone potential between the first
and second skimmer (parameters 7 and 8 in Figure
1) in the instrumental setup.18
In addition to the instrumental settings, differences
were also observed in the protein profiles from bacterial
cell extracts when the solvent conditions
were varied. Clearly, the experimental conditions
employed must be given due consideration for the
efficient detection of proteins or peptides in mixtures.
Moreover, the ability to vary the instrumental
settings rapidly means that we may hope to be
able to detect and analyze imperfectly separated
proteins/peptides on-line and in real-time for proteomic
applications. The tuning of instrumental
settings in the source and the source–analyzer interface
is thus likely to provide an important tool for
developing strategies for the efficient analysis of
proteins or peptides in mixtures, eventually
enabling wider proteome coverage than is possible
with current technology.
References
-
Smith, R.D. Trends in mass spectrometry instrumentation
for proteomics. Trends Biotechnol. 2002, 20,
S3–7.
- Lee, S.W.; Berger, S.J.; Martinovic, S.; Pasa-Tolic, L.;
Anderson, G.A.; Shen, Y.F.; Zhao, R.; Smith, R.D.
Direct mass spectrometric analysis of intact proteins of
the yeast large ribosomal subunit using capillary
LC/FTICR. Proceedings of the National Academy of
Sciences of the United States of America2002, 99, 5942–7.
- Aebersold, R.; Mann, M. Mass spectrometry-based
proteomics. Nature2003, 422, 198–207.
- Hayter, J.R.; Robertson, D.H.L.; Gaskell, S.J.;
Beynon, R.J. Proteome analysis of intact proteins
in complex mixtures. Molec. Cell. Proteom. 2003,
2, 85–95.
- Reid, G.E.; McLuckey, S.A. “Top down” protein characterization
via tandem mass spectrometry. J. Mass
Spec. 2002, 37, 663–75.
- Stephenson, J.L.; McLuckey, S.A.; Reid, G.E.; Wells,
J.M.; Bundy, J.L. Ion/ion chemistry as a top-down
approach for protein analysis. Curr. Opin. Biotechnol.
2002, 13, 57–64.
- Wysocki, V.H.; Resing, K.A.; Zhang, Q.; Cheng, G.
Mass spectrometry of peptides and proteins. Methods2005, 35, 211–22.
- Bogdanov, B.; Smith, R.D. Proteomics by FTICR mass
spectrometry: top down and bottom up. Mass
Spectrom. Rev. 2005, 24, 168–200.
- Wang, G.D. Electrospray Ionization Mass Spectrometry:
Fundamentals, Instrumentation & Applications; Cole,
R.B., Ed.; John Wiley & Sons: New York, 1997, pp
137–74.
- Chernushevich, I.V.; Verentchikov, A.N.; Ens, W.;
Standing, K.G. Effect of ion-molecule collisions in the
vacuum chamber of an electrospray time-of-flight
mass spectrometer on mass spectra of proteins. J.
Amer. Soc. for Mass Spectrom. 1996, 7, 342–9.
- Amad, M.H.; Cech, N.B.; Jackson, G.S.; Enke, C.G.
Importance of gas-phase proton affinities in determining
the electrospray ionization response for analytes
and solvents. J. Mass Spectrom. 2000, 35, 784–9.
- Iavarone, A.T.; Jurchen, J.C.; Williams, E.R. Effects of
solvent on the maximum charge state and charge state
distribution of protein ions produced by electrospray
ionization. J. Amer. Soc. for Mass Spectrom. 2000, 11,
976–85.
- Sterner, J.L.; Johnston, M.V.; Nicol, G.R.; Ridge, D.P.
Signal suppression in electrospray ionization Fourier
transform mass spectrometry of multi-component
samples. J. Mass Spectrom. 2000, 35, 385–91.
- Smith, R.D.; Loo, J.A.; Barinaga, C.J.; Edmonds,
C.G.; Udseth, H.R. Collisional activation and collision-activated dissociation of large multiply charged
polypeptides and proteins produced by electrospray
ionization. J. Amer. Soc. for Mass Spectrom. 1990, 1,
53–65.
- Belov, M.E.; Gorshkov, M.V.; Alving, K.; Smith, R.D.
Optimal pressure conditions for unbiased external ion
accumulation in a two-dimensional radio-frequency
quadrupole for Fourier transform ion cyclotron resonance
mass spectrometry. Rap. Commun Mass
Spectrom. 2001, 15, 1988–96.
- Schmidt, A.; Bahr, U.; Karas, M. Influence of pressure
in the first pumping stage on analyte desolvation and
fragmentation in nano-ESI MS. Anal. Chem. 2001,
73, 6040–6.
- Oberacher, H.; Walcher, W.; Huber, C.G. Effect of
instrument tuning on the detectability of biopolymers
in electrospray ionization mass spectrometry. J. Mass
Spectrom. 2003, 38, 108–16.
- Vaidyanathan, S.; Broadhurst, D.I.; Kell, D.B.;
Goodacre, R. Explanatory optimization of protein
mass spectrometry via genetic search. Anal. Chem.
2003, 75, 6679–86.
- Vaidyanathan, S.; Kell, D.B.; Goodacre, R. Selective
detection of proteins in mixtures using electrospray
ionization mass spectrometry: influence of instrumental
settings and implications for proteomics. Anal.
Chem. 2004, 76, 5024–32.
- Michalewicz, Z.; Fogel, D.B. How to Solve it: Modern
Heuristics. Springer-Verlag: Heidelberg, 2000.
Dr. Vaidyanathan is a Research Associate, School of Chemistry,
the University of Manchester, Sackville St., Manchester M60
1QD, U.K.; tel.: +44 0161 306 4414; e-mail:
[email protected].