An important fine chemical intermediate for pharmaceuticals, agrichemicals and polymers, 2,4,5-trifluorobenzoic acid (2,4,5-TFBA) is synthesized from tetrachlorophthalic anhydride via hydrolysis, fluorination and decarboxylation, respectively.10
As shown in Figure 1, the synthesis of 2,4,5-TFBA consists of five steps. It is essential in the laboratory and in industrial production to estimate the reaction time and analyze 2,4,5-TFBA; 2,4,5-TFBA has been detected and quantified using different methods, e.g., GC,11-13 GC/MS14-16 and capillary electrophoresis (CE).17 Use of HPLC for the synthesis of 2,4,5-TFBA has not been reported thus far.
 Figure 1 – Synthesis of 2,4,5-TFBA from tetrachlorophthalic anhydride.
Figure 1 – Synthesis of 2,4,5-TFBA from tetrachlorophthalic anhydride.The purpose of the present investigation was to use HPLC to track and control the synthesis of 2,4,5-TFBA; study the HPLC determination conditions for 3,4,6-trichlorophthalic acid, 3,4,6-trichloro-N-methylphthaldiamide, 3,4,6-trifluoro-N-methylphthaldiamide, 3,4,6-trifluorophthalic acid and 2,4,5-TFBA; and establish an accurate, sensitive and stable analytical method that is suitable for laboratory and industry.
Experimental
Reagents
Tetrachlorophthalic anhydride, ethyl acetate, sodium hydroxide, sulfolane, potassium fluoride, zinc powder, dimethyl sulfoxide (DMSO) and magnesium sulfate were of analytical reagent grade. The authors synthesized 3,4,6-trichlorophthalic acid; 3,4,6-trichloro-N-methylphthaldiamide; 3,4,6-trifluoro-N-methylphthaldiamide; 3,4,6-trifluorophthalic acid and 2,4,5-TFBA. Acetonitrile, trifluoroacetic acid and citric acid were of chromatographic grade. Ultrapure water was used.
Preparation of 2,4,5-TFBA
The 2,4,5-TFBA was prepared using the procedures given in the literature.10 To 750 mL of 10% sodium hydroxide, 75 g (0.262 mol) of tetrachlorophthalic anhydride and 102.18 g (1.572 mol) of zinc powder were added. The reaction mixture was then heated and stirred for 4 hr at a bath temperature of 60 °C. The mixture was cooled to room temperature, washed with sodium hydroxide and distilled water (2 × 100 mL), respectively, and filtered; 100 mL of ethyl acetate was added to the filter residue, and hydrochloric acid was then added until the filter residue dissolved. The aqueous layer was extracted with ethyl acetate (2 × 100 mL). The organic layer combined with the extracted ethyl acetate was dried with magnesium sulfate, filtered and evaporated to dryness in vacuo to give 60.2 g of 3,4,6-trichlorophthalic acid (mp 148–149 °C). The product yield was 85.2%.
To 50 g of 3,4,6-trichlorophthalic acid, 400 mL of acetic acid and 14.9 g of 40% aminomethane aqueous solution were added. The reaction mixture was then heated and stirred for 6 hr at a bath temperature of 65 °C. The mixture was cooled to room temperature and filtered. The filter residue was washed with ice water (5 × 100 mL) and dried in vacuo to give 49 g of 3,4,6-trichloro-N-methylphthaldiamide (mp 164–166 °C). The product yield was 99.5%.
To 20 g of 3,4,6-trichloro-N-methylphthaldiamide, 100 mL of sulfolane and 50 mL of toluene were added. The reaction mixture was heated to reflux until there was no water. Next, 19.7 g (0.34 mol) of anhydrous potassium fluoride was added to the reaction mixture, and under the protection of nitrogen the mixture was stirred at 240 °C for 2 hr. The mixture was cooled to room temperature and poured into 500 mL of ice water. Immediately, a large number of solids was produced. The produced solids were dried in vacuo to give 14.3 g of 3,4,6-trifluoro-N-methylphthaldiamide (mp 175–176°C). The product yield was 87.8%.
To 10.0 g of 3,4,6-trifluoro-N-methylphthaldiamide, 100 mL of 25% sulfuric acid was added. The mixture was stirred at 125 °C for 4 hr, and was then cooled to room temperature and extracted with ethyl acetate (4 × 100 mL). The extracted ethyl acetate was dried with magnesium sulfate, filtered and evaporated to dryness in vacuo to give 7.9 g of 3,4,6-trifl uorophthalic acid (mp 157–158 °C). The product yield was 77.1%.
A 100-mL single-neck flask equipped with a condenser and magnetic stirrer was charged with 10.0 g of 3,4,6-trifluorophthalic acid and 100 mL of DMSO. The reaction mixture was stirred at 149 °C for 28 hr. The flask was then allowed to cool to room temperature and the contents were poured into 400 mL of water and extracted with 4 × 100 mL of ethyl acetate. The combined organic extracts were washed with water (2 × 100 mL), dried with magnesium sulfate, filtered and evaporated to dryness in vacuo to give the crude product. The pure product (7.7 g of 2,4,5-TFBA, mp 101–102 °C) was obtained by column chromatography on silica gel of the crude product. The product yield was 96.3%. The product was properly characterized by 1H NMR and GC/MS. The total yield of the five steps reaction was 55.5%.
Representative data
- 2,4,5-TFBA: 1H NMR (CDCl3, 500 MHz, δ ppm): 7.27 (s, 1H), 7.96 (s, 1H), 10.92 (s, 1H)
- GC/MS (m/z): 176(M+), 159, 131,112, 81.
HPLC system
A Dionex P680 HPLC system (Thermo Fisher Scientific, Waltham, Mass.) was equipped with a P680A programmable pump, UVD 170U detector and 7725 manual injector. An ODS-C18 (4.6 × 150 mm, 5 μm) column from Supelco (Bellefonte, Penn.) was used. The chromatographic system was controlled using Dionex (Sunnyvale, Calif.) Chromeleon chromatography manager software (version 6.80).
Preparation of standard solutions
Standard solutions of 3,4,6-trichlorophthalic acid; 3,4,6-trichloro-N-methyl-phthaldiamide; 3,4,6-trifluoro-N-methylphthaldiamide; 3,4,6-trifluorophthalic acid and 2,4,5-TFBA were prepared by dissolving an accurately weighed quantity (about 15 mg) of 3,4,6-trichlorophthalic acid; 3,4,6-trifluorophthalic acid and 2,4,5-TFBA in a mixture of acetonitrile: water:trifluoroacetic acid (40:60:2), respectively, and 3,4,6-trichloro-N-methyl-phthaldiamide; 3,4,6-trifluoro-N-methylphthaldiamide in a mixture of acetonitrile:0.1% citric acid aqueous solution (55:45), respectively, and adjusted to the l00-mL mark. The solutions were analyzed directly by HPLC.
HPLC operating conditions
The mobile phase was a mixture of acetonitrile:water:trifluoroacetic acid (40:60:2) and a mixture of acetonitrile:0.1% citric acid aqueous solution (55:45) with a flow rate of 1.0 mL/min. The samples were kept at room temperature before HPLC and were filtered and degassed before use. Detection wavelength was 272 nm. Each sample was filtered by a 0.45-μm filter and injected 5 μL each time. All chromatographic analyses were performed isocratically and at room temperature.
Results and discussion
In order to apply the HPLC method in the synthesis of 2,4,5-TFBA, the HPLC determination conditions were first studied for 3,4,6-trichlorophthalic acid; 3,4,6-trichloro-N-methyl-phthaldiamide; 3,4,6-trifluoro-N-methylphthaldiamide; 3,4,6-trifluorophthalic acid and 2,4,5-TFBA. The Dionex P680 HPLC system was equipped with a UVD 170U detector, which can detect samples at four detection wavelengths simultaneously. The 3,4,6-trichlorophthalic acid; 3,4,6-trichloro-N-methyl-phthaldiamide; 3,4,6-trifluoro-N-methyl-phthaldiamide; 3,4,6-trifluorophthalic acid and 2,4,5-TFBA were detected at 240, 254, 272 and 280 nm, respectively. The wavelength (nm) max. absorbances of the five samples were all 272 nm; thus 272 nm was chosen as the detection wavelength.
The 3,4,6-trichlorophthalic acid; 3,4,6-trichloro-N-methyl-phthaldiamide; 3,4,6-trifluoro-N-methylphthaldiamide; 3,4,6-trifluorophthalic acid and 2,4,5-TFBA were detected at a flow rate of 0.5, 1.0 and 1.5 mL/min, respectively. The retention time of the five samples varied from 8 min to 20 min at a flow rate of 0.5 mL/min; the detection time was longer. The retention time of five samples is too short not to be separated thoroughly, and the peaks overlap at a flow rate of 1.5 mL/min. Thus the flow rate of 1.0 mL/min was chosen for good separation effect.
At the HPLC operation conditions described above, the retention time was 11.438 min, 3.781 min and 5.154 min for 3,4,6-trichlorophthalic acid (Figure 2); 3,4,6-trifluorophthalic acid (Figure 3) and 2,4,5-TFBA (Figure 4), respectively, using a mixture of acetonitrile:water:trifluoroacetic acid (40:60:2) as mobile phase. The retention time was 11.108 min and 11.528 min for the 3,4,6-trichloro-N-methylphthaldiamide (Figure 5) and 3,4,6-trifluoro-N-methylphthaldiamide (Figure 6) using a mixture of acetonitrile:0.1% citric acid aqueous solution (55:45) as mobile phase. The content of the five samples with area normalization method was 96.40%, 100%, 95.00%, 97.10% and 99.69% for the 3,4,6-trichlorophthalic acid; 3,4,6-trichloro-N-methylphthaldiamide; 3,4,6-trifluoro-N-methyl-phthaldiamide; 3,4,6-trifl uorophthalic acid and 2,4,5-TFBA, respectively.
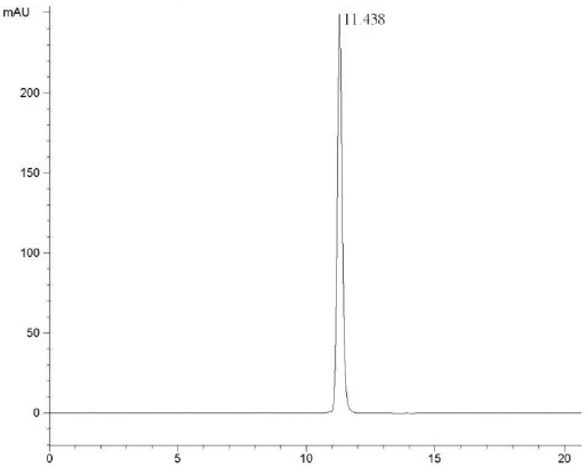 Figure 2 – Chromatogram of 3,4,6-trichlorophthalic acid.
Figure 2 – Chromatogram of 3,4,6-trichlorophthalic acid.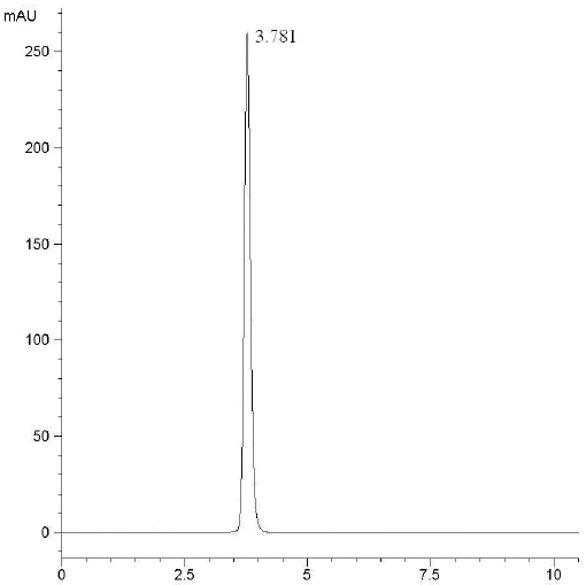 Figure 3 – Chromatogram of 3,4,6-trifluorophthalic acid.
Figure 3 – Chromatogram of 3,4,6-trifluorophthalic acid.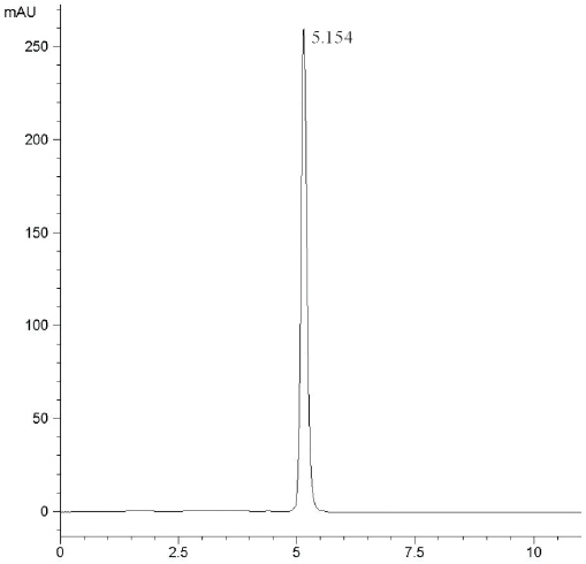 Figure 4 – Chromatogram of 2,4,5-TFBA.
Figure 4 – Chromatogram of 2,4,5-TFBA.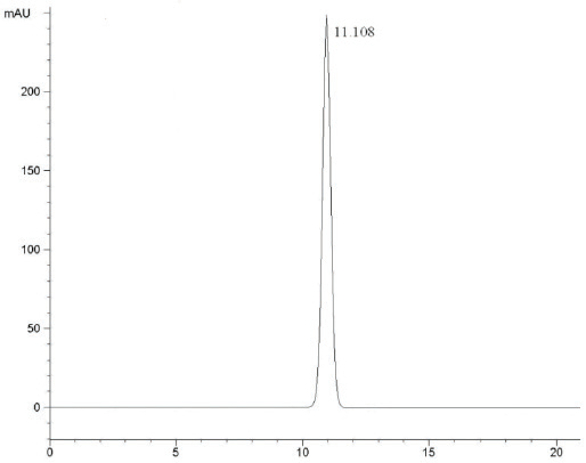 Figure 5 – Chromatogram of 3,4,6-trichloro-N-methylphthaldiamide.
Figure 5 – Chromatogram of 3,4,6-trichloro-N-methylphthaldiamide.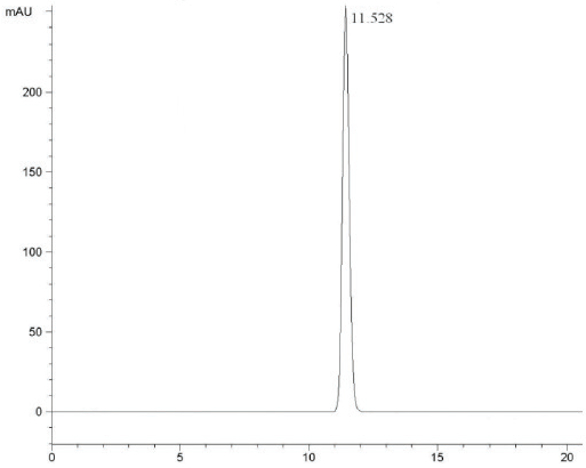 Figure 6 – Chromatogram of 3,4,6-trifluoro-N-methylphthaldiamide.
Figure 6 – Chromatogram of 3,4,6-trifluoro-N-methylphthaldiamide.On this basis, the HPLC method was used for the synthesis of 2,4,5-TFBA for the first time. The reaction solutions of five reaction steps such as the third step and the fifth step in the synthetic process were analyzed by HPLC, respectively.
Conclusion
For the first time, HPLC with UV detection was studied for the detection and quantification of 3,4,6-trichlorophthalic acid; 3,4,6-trichloro-N-methylphthaldiamide; 3,4,6-trifluoro-N-methylphthaldiamide; 3,4,6-trifluorophthalic acid and 2,4,5-TFBA. HPLC can be used to estimate the reaction time in the synthesis of 2,4,5-TFBA. The mobile phase is a mixture of acetonitrile:water:trifluoroacetic acid (40:60:2) and acetonitrile:0.1% citric acid aqueous solution (55:45). The two types of mobile phase have only three components that are easily made up. The detection wavelength was 272 nm. The method is accurate, sensitive, stable, reproducible and suitable for routine analysis.
References
- Takaoki, K.; Miyatake, T. EP 2048167; 2009.
- Takaoki, K.; Miyatake, T. EP 2048168; 2009.
- Takaoki, K.; Miyatake, T. EP 2048170; 2009.
- Erich, K; Uwe, P. et al. US 5200548; 1993.
- Erich, K; Uwe, P. et al. US 5777181; 1998.
- Xiu, W.; Panos, C.K. et al. US 5576455; 1996.
- Xiu, W.; Panos, C.K. et al. US 5633399; 1997.
- Xiu, W.; Panos, C.K.et al. US 5637765; 1997.
- Ming-quan, Zh.;Guo-ming, Y. et al. Chin. J. Pharm. 2004, 35, 714–15.
- O’Reilly, N.J.; Derwin, W.S. et al. US 5233085; 1993.
- O’Reilly, N.J.; Derwin, W.S. et al. EP 0431294A2; 1991.
- O’Reilly, N.J.; Derwin, W.S. et al. US 4935541; 1990.
- Qi, J.S. US 5336806; 1994.
- Horst, S. US 5093529; 1992.
- Karsten, M.; Andreas, S. J. Chromatogr. A 2012, 1260, 9.
- Tsuguhide, I.; Fumihiko, K. et al. J. Sep. Sci. 2009, 32, 381.
- Hugues, P.; Coralie, S.P. et al. US 20110260051; 2011.
The authors are with the School of Chemistry and Chemistry Engineering, Lingnan Normal University, Zhanjiang 524048, P.R. China; tel.: +86 0759 3238076; fax: +86 07593183510; e-mail: [email protected].