Modern bioanalytical laboratories are tasked with providing high-quality analytical results from complex biological samples in a high-throughput environment. These needs have to be balanced with the demands of the business, where compliance to legislation, improved efficiency, and reduced cost are major considerations.
The burden on the bioanalyst is exacerbated by the move to higher-efficacy drugs and long-acting formulations that reduce the maximum concentration reached by a drug in the body and consequently the required quantitation limits. This is further compounded by the desire to take advantage of the replacement, refinement, and reduction policy that is driving the industry toward reduced animal model use.
In order to meet these demands, bioanalytical solutions need to be:
- Robust and rugged
- Highly reproducible
- Able to produce high levels of extract cleanliness
- Highly sensitive
- Cost effective.
Despite these requirements, analytical failures do occur that result in time-consuming and costly reanalysis. A rugged and robust assay is only possible when the analytical tools at your disposal are produced to a high standard of uniformity and quality. The choice of sample preparation products is important since this is a high-risk area of the method and there is a significant chance of analytical failure. Analytical failures often arise due to inherent problems with the design of classical solid-phase extraction (SPE) devices that affect assay performance.
Conventional solid-phase extraction
Conventional SPE cartridges and plates are packed with a loose powder (typically polymeric material or silica) positioned between two frits. During the production process, it is possible to over- or underfill the holder with the required amount of material; once packed, these beds are prone to settling and voiding during production and transport (see Figure 1).
 Figure 1 – Examples of conventional SPE product issues.
Figure 1 – Examples of conventional SPE product issues.These packing inconsistencies, particularly channeling and voiding, affect assay ruggedness due to variable compound recovery during sample preparation, which manifests as poor analyte response reproducibility.
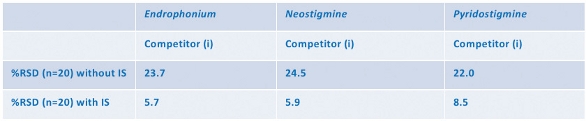
This is illustrated in the example in Figure 2 and Table 1. Twenty aliquots of the same aqueous solution containing an anesthetic and two reversal agent compounds underwent a standard extraction procedure on a competitor loose-packed material. The data show the response with and without internal standard (IS) compensation.
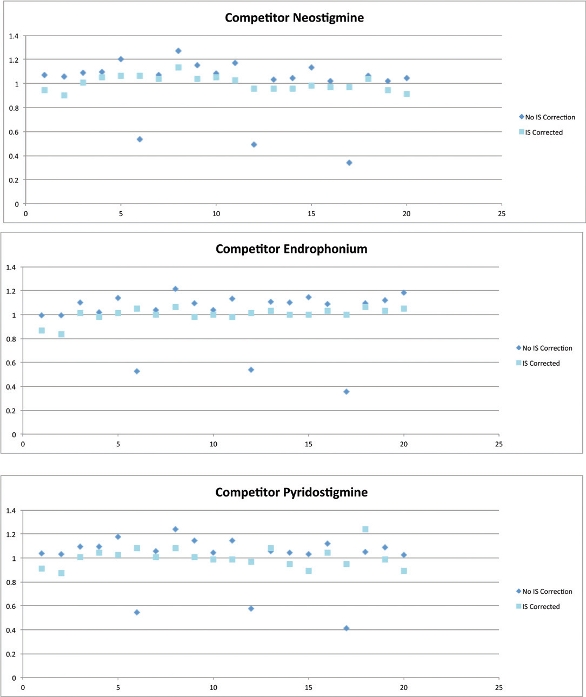 Figure 2 – Loose-packed competitor results for three representative compounds.
Figure 2 – Loose-packed competitor results for three representative compounds.Table 1 – Reproducibility with and without IS correction
Looking at the data without IS, it is clear that most of the results demonstrate a variance that is within acceptable experimental error; however, three of the results show a far lower response for all three analytes (the response drops to around 50% of the baseline result). In these cases, the compound recovery has been compromised. This can be attributed to a number of different causes: operator error during the process, sample losses, instrument malfunction, or issues within the sample preparation procedure, to name a few.
However, based on these data, is it fair to say that variations in recovery do not matter if we use an IS? An internal standard is generally employed to compensate for assay variability, which presents as variation in peak response. Variation is often due to inconsistent sample recovery during extraction or as a result of matrix effects associated with poorly cleaned complex (biological) samples.
Figure 2 shows that once the samples are internal standard corrected, the low recoveries are not obvious. The internal standard recovery is compromised to the same amount as the analyte of interest, and peak area ratio is maintained, giving the illusion of acceptable reproducibility.
Therefore, use of an internal standard can (and is actually designed to) mask method variability. Often the internal standard does this effectively, and the analyst can be confident in the quality of the results; however, some issues cannot be resolved. In particular, reduced peak height and area due to lower recovery can be an issue.
A recent draft legislation by the FDA1 indicates that the internal standard should be monitored for drift and written into standard operating procedures (SOPs). Monitoring of these data provides an indication of where sample failure occurs and is indicative of the reproducibility of the assay.
Sample recovery
What happens when sample recovery is compromised during extraction at or near the quantitation limit of the assay? In this scenario, analyte loss during extraction, which leads to reduced peak heights, may result in the sample peak not being detected. In this case, quantification at low concentration becomes impossible and potentially causes batch failure.
This can be seen in the three chromatograms in Figure 3, where a single concentration standard results in two very different peak responses (and S/N). This occurs for all three compounds for a given sample, suggesting it is an extraction issue.
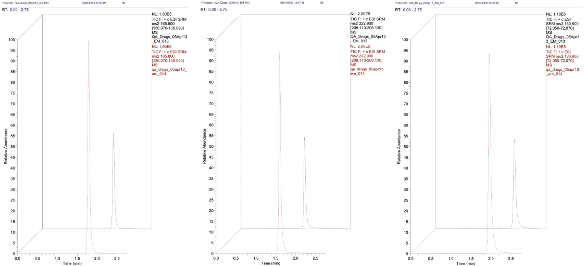 Figure 3 – Chromatograms showing variation in response.
Figure 3 – Chromatograms showing variation in response.So why don’t we see reproducibility issues with our sample preparation technique? This is an important question to ask and the simplest answer is that we don’t always look for it. How often do we go back and critically assess the reasons for batch failure when there is not an immediately obvious cause like instrument failure or obvious operator error? When we do, it is not always easy to tease out the root cause of the issue, and in the modern analytical laboratory often we don’t have the time.
Alongside this, it is fair to say that in general SPE product quality is acceptable and not every cartridge or plate shows symptoms of voiding, channeling, or inaccurate bed weight. However, SPE product quality can vary from individual cartridge to cartridge, box to box, or batch to batch, and it is often obvious if you look for it.
Even though one plate may provide consistent results, there is no guarantee that the next cartridge or plate will. This can be seen in Figure 4, where three loose-packed 96-well SPE plates from three different media lots were investigated under “real life” experimental conditions. An analyte, rosuvastatin, at a physiologically relevant concentration (10 ng/mL) in human plasma was taken through a standard extraction protocol on a reversed-phase SPE cartridge and the peak area was plotted for each of the 216 data points (72 per plate).
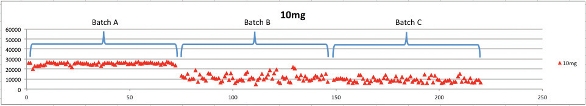 Figure 4 – Three different lots of competitor loose-packed SPE plates for rosuvastatin in human plasma at a single concentration.
Figure 4 – Three different lots of competitor loose-packed SPE plates for rosuvastatin in human plasma at a single concentration.The data clearly demonstrate the variability in product performance that may occur with conventional loose-packed SPE products. The difference in recovery between the first and subsequent two plates is clear. Plate one gives very reproducible, high-recovery results, but the other two plates fail to reproduce this.
The obvious impact of regulation on assay reproducibility is the requirement for assay validation. Precision and accuracy at multiple levels are well understood during assay validation, but beyond this, how often do we look at multiple SPE lots during this process and, more importantly, do we consider looking at the data without internal standard compensation? Realistically, should we worry?
Recent publications by the EMEA,2 the regulatory authority for Europe, have started to appreciate that high levels of analytical variability can have a negative influence on the quality of the data. Specifically, the EMEA guidelines now state that the calculated matrix factor imprecision for the internal standard from six different lots of matrix should not exceed 15%.
Essentially, employing an internal standard should make the data look better, but inherently poor data are poor data, and it appears that this is now, more than ever, a focus area. The ability of an analytical method to repeatedly give the same result for a given sample is essential, and Incurred Sample Reanalysis (ISR) is now used to determine this. ISR is designed to test the robustness and reproducibility of a method; the more robust and reproducible the method, the less likely it is to fail.
Among the issues ISR is designed to highlight are:
- Sample processing error
- Impact of changes during sample preparation
- Change in laboratory environment.
All of these will be impacted by the choice of sample preparation device.
The introduction of Incurred Sample Reanalysis and the Internal Standard Compensated Matrix Factor by regulatory authorities suggests that the influence of the matrix on assay reproducibility will become more important in the future for assay validation.
Sample preparation techniques for bioanalysis are required first and foremost to remove unwanted matrix components and interferences from the final extract solution. This is of particular importance when using atmospheric pressure ionization mass spectrometry (API–MS) for analyte detection, where the presence of matrix impurities may cause modification of ionization.
The most obvious consequence of choosing different extraction protocols (for example, dilute and shoot, protein precipitation, solid supported liquid extraction, or SPE) is in the cleanliness of the final extract. As can be seen in Figure 5, leachable and extractable materials from the sample cleaning device itself also have an impact on extract cleanliness, and much of the effort given to removing matrix interferences may be wasted if these compounds are present.
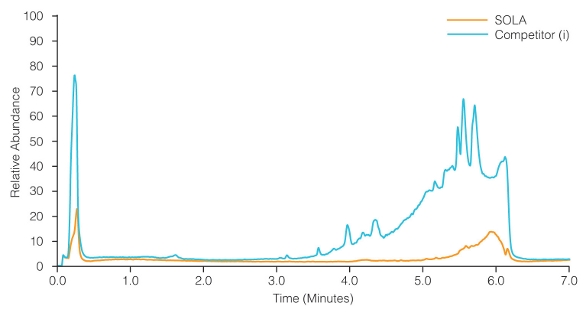 Figure 5 – Dichloromethane extraction from the sample cleaning device.
Figure 5 – Dichloromethane extraction from the sample cleaning device.Due to their nature, biological matrices such as plasma present a difficult challenge in obtaining reproducible results. This is not only due to the complex nature of the matrix, but to the fact that matrix content changes with time in a single individual, from person to person, with disease state and for a myriad number of other reasons. Due to this, it is important to remove as much of the matrix as possible during sample extraction.
A major requirement for modern analytical methods is high sensitivity, or improved limits of detection and quantitation. It is often assumed that the simplest way to improve sensitivity is with enhancements to the detection technique. While this is effective, it may not be the most expedient or most cost-effective way to improve assay sensitivity. Often, increasing the concentration step in the sample extraction procedure can give improvements in compound sensitivity. However, with classical loose-packed SPE there have always been inherent problems when doing this.
The frits, which are present in traditional SPE products to contain the powdered material, need to be porous. Unfortunately, this porosity increases the total volume within the frits and adds holdup (or dwell) volume to the SPE product. The effects of this holdup volume become more pronounced as the elution volume decreases, leading to reduced response and decreased reproducibility, as can be seen in Figure 6.
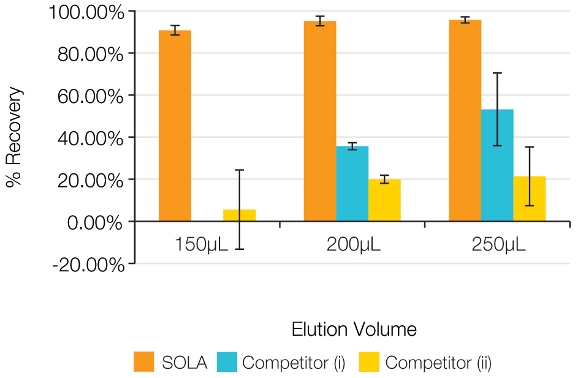 Figure 6 – Reproducibility and recovery levels at reduced elution volumes.
Figure 6 – Reproducibility and recovery levels at reduced elution volumes.New SPE design and improved performance
Until recently there has been no alternative to the conventional loose-packed SPE design. SOLA SPE products (Thermo Fisher Scientific, Runcorn, Cheshire, U.K.) provide a step change in design and product performance. SOLA was developed especially to meet the needs of the bioanalyst.
The proprietary SOLA manufacturing process generates an SPE product that eliminates the issues with conventional loose-packed SPE. By combining the support material and active media components into a solid uniform sorbent bed, the need for frits is eliminated (Figure 7).
 Figure 7 – SOLA’s proprietary design removes channeling, voiding, and bed weight differences.
Figure 7 – SOLA’s proprietary design removes channeling, voiding, and bed weight differences.Stable and controllable flow through the SPE device is another key factor controlling reproducibility of the final analytical method. This is especially important in low-bed-weight devices in which flow control is more difficult due to lower backpressure from the sorbent. Carrying out ion exchange methods where the kinetics are known to be slower also requires more controlled flow. These problems are exacerbated in biological samples due to their viscosity and inherent variability.
In conventional loose-packed devices, the macroporosity of the SPE product (i.e., flow between the porous particles) is defined by the pressure applied to the frits during production: the greater the pressure, the more tightly packed the device, which results in more resistance to flow. In addition, flow resistance is affected by the presence of fines in the loose powder, the particle size distribution of the media, and the tendency for some polymeric media to swell upon wetting.
SOLA is manufactured with high-quality polymeric media that is sized to remove fines and minimize particle size distribution and is highly cross-linked to reduce swelling. Additionally, the macroporous structure is defined by the well-controlled, reproducible manufacturing process, resulting in uniformity cartridge to cartridge or well to well, plate to plate, and batch to batch. Flow characteristics can be measured by a simple airflow experiment that is used as a quality control check during production (see Figure 8).
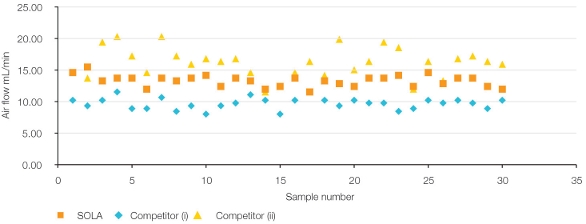 Figure 8 – Flow-through characteristics of SOLA SPE and conventional loose-packed 10-mg SPE.
Figure 8 – Flow-through characteristics of SOLA SPE and conventional loose-packed 10-mg SPE.Analytical reproducibility is fundamental to the success of bioanalytical studies. SOLA’s robust design removes the risks associated with conventional loose-packed SPE products, which can cause failure and costly reanalysis. SOLA helps ensure the reproducibility required for work flow success by providing:
- No voiding
- No channeling
- A cleaner product
- Improved flow profile.
This can be seen in Figure 9 and Table 2. The graph shows data for LC-MS/ MS response of the same three compounds extracted using a competitor (i) conventional loose-packed product in Figure 2.
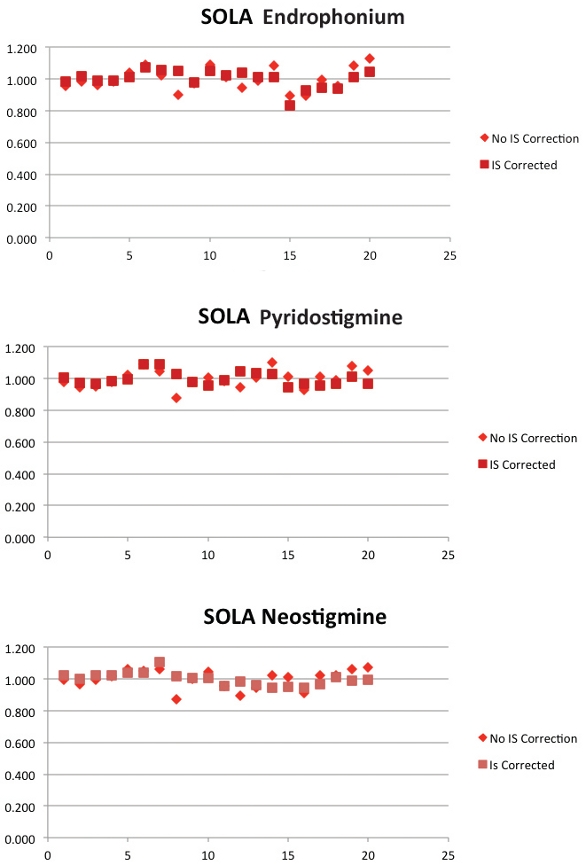 Figure 9 – SOLA results for three representative compounds.
Figure 9 – SOLA results for three representative compounds.Table 2 – Reproducibility with and without IS correction
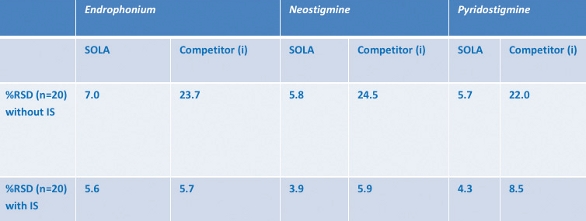
Variation in recovery has been calculated as a percentage relative standard deviation. SOLA shows a minimal difference between reproducibility results with and without an internal standard (<2%), whereas the competitor (i) conventional loose-packed SPE product shows significantly worse performance when IS was removed, suggesting well-to-well differences in recovery and an inherent variability in the competitor product (Table 2).
SOLA demonstrates good intra-lot variability with the added benefit of consistency well to well, plate to plate, and lot to lot (see Figure 10). In this figure, the same analysis was carried out over three 96-well plates from three separate lots of material.
 Figure 10 – Three different lots of SOLA SPE plates for extraction of rosuvastatin in human plasma at a single concentration.
Figure 10 – Three different lots of SOLA SPE plates for extraction of rosuvastatin in human plasma at a single concentration.In addition, the use of high-quality raw materials coupled with the proprietary manufacturing process provides a cleaner product with reduced leachables and extractables. This is shown in Figure 5, where SOLA products and competitor (i) conventional loose-packed SPE products have both been exposed to dichloromethane subsequently analyzed by LC-MS.
SOLA also allows lower solvent volumes. Why is this important to your work flow? The ability to elute a sample in a lower solvent volume and use less solvent during the SPE process means:
- Reduced solvent cost
- Reduced processing time
- Concentrated extract.
This provides significant advantage for high-throughput bioanalytical laboratories, where improved efficiencies combined with greater confidence in results are highly desirable.
SOLA achieves this due to its fritless design, which reduces holdup volumes associated with frits within conventional loose-packed SPE products. Figure 6 shows that SOLA products achieve very high recovery levels, even with low volumes of extract solvents, resulting in a more concentrated analyte and increased sensitivity. These low-volume extractions would be significantly compromised when using a conventional loose-packed, low-bed-weight SPE product.
Conclusion
The choice of SPE product can have a significant effect on the throughput and cost effectiveness of the bioanalytical work flow. SOLA SPE technology can mitigate this by providing greater reproducibility combined with low elution volumes, lower failure rates, and cleaner extracts, which are key to driving improvements in productivity.
References
- Guidance for Industry, Bioanalytical Method Validation, Draft Guidance, U.S. Department of Health and Human Services Food and Drug Administration, Sept 2013.
- Guideline on Bioanalytical Method Validation, European Medicines Agency, Jul 21, 2011.
Mike Oliver is Sample Preparation Product Manager, Tim Liddicoat is Support Manager, and Ken Meadows is Senior Application Scientist, Chromatography and Consumables, Thermo Fisher Scientific, Tudor Rd., Manor Park, Runcorn, Cheshire WA7 1TA, U.K.; tel.: +44 (0) 1928 534335; fax: +44 (0) 1928 588106; e-mail: [email protected].