Part 1 of this two-part paper explains the basic principles of evaporation and concentration, and outlines some of the commonly used technologies. Part 2 (https://www.americanlaboratory.com/914-Application-Notes/131586-Understanding-Concentration-and-Evaporation-Technology-Part-2-Latest-Developments-in-System-Technology/) reviews the wide variety of systems used in evaporation, including pumps, cold traps, and evaporators.
Solvent removal is an essential process across a broad range of applications in the pharmaceutical, chemical, and biotechnology industries. Although a diversity of sample formats and solvents are used, no single solvent removal technique provides a universal solution. Various commercial evaporation and concentration systems have been developed to accommodate the range of applications. These systems and associated hardware‒vacuum pumps, cold traps, and heating technologies‒have recently benefitted from exciting developments in freeze-drying and centrifugal concentration technologies, enabling enhanced evaporation performance and improved sample integrity. Further, advances with new-generation high-power cold traps are offering improved solvent recovery, thereby reducing the environmental impact of the evaporation/concentration process.
An understanding of the most up-to-date evaporation and concentration processes, their application at a practical level, and use of the latest equipment permits optimization of protocols for improved and more rapid sample concentration.
Processes involved with solvent removal
During solvent removal, energy is applied as heat such that the liquid is vaporized to gas, which is removed to leave a concentrated or solvent-free (dry) product. Many systems are referred to generically as “evaporators.” However, true evaporation is vaporization to gas at the liquid surface; for many “evaporators,” boiling occurs rather than evaporation. The process of freeze-drying involves neither evaporation nor boiling, but sublimation, that is, a shift from solid to vapor phase without a liquid phase.
The phase of a substance is determined by two major factors‒heat and pressure‒and the temperature at which boiling or vaporization occurs is set by the pressure. Therefore, vacuum concentrators apply vacuum in the system to decrease a solvent’s boiling point so that liquid vaporization occurs at lower temperatures, e.g., water boils at –7.5 °C at 10 mbar pressure. Similarly, in freeze dryers, heat energy supplied to a frozen sample at low pressure transfers sufficient energy for thawing, but the pressure is insufficient for liquid formation, and hence the solvent sublimes to gas. Generated vapor is removed by a cold trap or condenser, where solvent is recovered.
Heat and temperature
Solvent removal systems use an input of heat energy to induce solvent vaporization, and various heating mechanisms are employed, such as electrical heating blocks, lamps, or low-temperature steam. Heat and temperature, although linked, are different, and it is important to distinguish between them. Heat refers to heat energy measured in Joules, whereas temperature measures the level of heat energy, i.e., the hotness or coldness of an object. Samples referred to as heat-sensitive are typically temperature-sensitive, and a majority of samples can be heated without degradation, provided the temperature remains within defined boundaries. Applying vacuum in a system decreases a solvent’s boiling point so that liquid vaporization occurs at lower temperatures that are safe for the sample.
Heat and temperature are related by the equation Q = mcΔT, where Q is heat energy added, m is the mass of the object, c is the specific heat capacity of the heated object, and ΔT is the change in temperature. ΔT can be expressed in terms of heat added as ΔT = Q/mc. These equations hold true when all other parameters remain the same. However, at a change in phase, added heat energy does not increase temperature since energy is required for the change in state, for example, from liquid to gas. Therefore, in true evaporative systems (no boiling), a sample is at the temperature of the system controlling it, whereas in a freeze dryer that actively freezes products, the sample is at the temperature at which it is frozen, and then its temperature is governed by the ice sublimation temperature controlled by the vacuum level.
 Figure 1 – Relationship between pressure and boiling point for some common solvents.
Figure 1 – Relationship between pressure and boiling point for some common solvents.The opposite dynamic exists in vacuum concentrators that boil solvent. When a sample is wet and boiling, the sample is at the boiling temperature of the liquid. Figure 1 shows the relationship between boiling point and pressure for some common solvents. At this stage, it is possible to heat the system to high temperatures, and a sample will not reach this temperature until the solvent is completely removed. Only when all solvent is removed will a sample warm to the temperature of the system. Accurate control and monitoring of sample temperature are therefore critical. Control of the sample holder temperature typically offers effective protection, since the sample within cannot exceed this temperature unless the sample is heated directly and independently of the holder. This necessitates using solid metal holders constructed from materials such as aluminum that can transmit the maximum amount of heat into the sample and are cooled by evaporating solvent. Real-time temperature monitoring is built into some systems, allowing accurate determination of when evaporation is complete and thereby avoiding overheating of samples.
Drying methods
Freeze-drying
Freeze dryers are available as two basic types: 1) a system that actively freezes samples placed on chilled shelves similar to a laboratory freezer, and 2) (passive) systems that do not actively freeze but instead utilize a manifold with attached flasks that contain sample either directly or within vials. A high vacuum is often used so that samples remain frozen and therefore well-preserved as solvent sublimes and is collected in the cold trap.
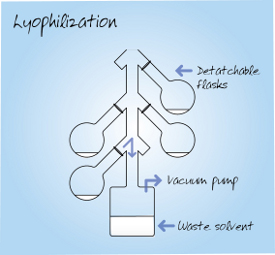
Figure 2 – Lyophilization process.
A typical freeze-dried product is a diffuse “fluffy” powder that has a very high level of dryness (due to the large surface area available for solvent removal) and is easy to weigh and redissolve. Some samples, such as DNA, may require careful handling during movement to avoid loss of the fine powder. Freeze-drying is a comparatively slow batch process, although a range of configurations are available that can accommodate large batches of samples per cycle. Solvent bumping may occur, but this can be reduced by prefreezing samples when feasible. The freezing process limits the technique to aqueous solutions or one of a few simple organic solvents that freeze easily, e.g., tertiary butanol or 1,4-dioxane. Samples containing volatile solvents must be actively frozen at very low temperatures, which may demand vacuum control at very low pressure and are so cold that the condenser does not function efficiently. The freeze-drying, or lyophilization, process is illustrated in Figure 2.
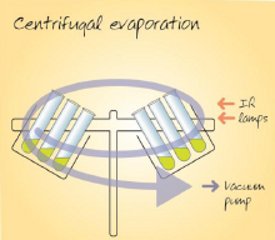
Figure 3 – Centrifugal evaporation process.
Centrifugal concentration
Centrifugal concentrators induce solvent boiling under vacuum and thus samples are cold; however, in contrast to freeze dryers, the samples are not frozen, and so the process can be faster than freeze-drying. Care must be taken with centrifugal evaporation of aqueous samples that are prone to freezing. Centrifugal evaporators use cold traps to recover the vaporized solvent. Centrifugation ensures that solvent boils from the sample surface downwards (see Figure 3), thereby minimizing boiling over and solvent bumping and preventing sample loss and cross-contamination. Solvent at the liquid surface is at the pressure of the equipment, whereas solvent below this level is at higher pressure due to the extra weight of solvent multiplied by the g force exerted by the centrifuge rotor. Systems with very high rotor speeds generating 500g or more are proven to prevent solvent bumping. The centrifugal evaporation technique accommodates a wide range of solvents and can concentrate, dry to a film, or freeze-dry samples.
Advanced centrifugal evaporation systems can achieve rapid freeze-drying by concentrating the majority of a larger volume before freeze-drying the final few milliliters of sample. Samples must be processed in batches, although large numbers of small samples can be processed at the same time. Early systems were viewed as slow, but recent developments in high-energy sample heating using low-temperature low-pressure steam, such as employed by the Genevac Rocket™ Evaporator (Genevac, Suffolk, U.K.), enable rapid concentration of larger volumes at the speed of a rotary evaporator. Such systems operate without solvent bumping and the consequent need for continual monitoring associated with rotary evaporators.
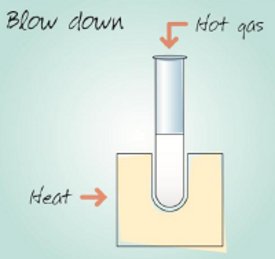
Figure 4 – Blow-down evaporation process.
Blow-down evaporation
In these evaporator systems, an inert gas such as nitrogen is blown down through needles onto samples in tubes, vials, or microplates to create a flow over the liquid surface (see Figure 4). This alters the equilibrium between the vapor and liquid phases to favor the vapor phase. Heat is normally applied to the samples to hasten evaporation, and prewarmed gases can also be used. The technique enables open-access equipment use, and is comparatively inexpensive, ranging from self-assembled apparatus to simple commercial systems. Although blow-down evaporation is relatively fast for volatile solvents, it can be slow for solvents with high boiling points or those that are difficult to evaporate such as water. Blow-down evaporation samples are prone to heating up during the process, since they are at the temperature of the heating block or bath during evaporation, and consequently the technique offers poor recovery of volatile analytes. As a manual process, blow-down evaporation requires continuous monitoring by the user to detect the endpoint of the drying process. The technique typically achieves poor final dryness and splashing may occur, particularly if the gas flow rate is too high, leading to sample cross-contamination. A common use of blow-down is for concentrating large volumes to just a few milliliters for subsequent processing by other techniques. Specialized teat-ended tubes are available for this procedure, and some commercial systems have basic automatic stop mechanisms based on liquid level detection.
Vortex evaporation and rotary evaporation
 Figure 5 – Vortex evaporation process.
Figure 5 – Vortex evaporation process.Vortex evaportors boil batches of samples under vacuum to keep the samples cold throughout vaporization, while swirling the sample tubes to create a vortex (see Figure 5). A rotary evaporator is essentially similar, but is for a single sample contained in a flask (see Figure 6). The vortex created generates a large sample surface area for evaporation, making the process relatively fast. However, the resultant dried product is spread across the vessel walls, which can make sample recovery more difficult. Moreover, in contrast to centrifugal concentrators, the swirling movement generates insufficient g force to prevent solvent bumping, and thus vortex evaporators are prone to sample loss and cross-contamination. In some vortex systems, evaporation is further accelerated by the use of heating lamps directed into the sample tubes, but these systems are prone to overheating all or part of a sample as it dries.
Factors affecting the speed of evaporation
Three factors affect the speed of concentration: heat energy supply, vapor removal, and solvent surface area. For boiling solvents, the faster heat energy is supplied, the faster the solvents boil. Similarly, for evaporative systems, more heat equals faster evaporation, although samples are at the set system temperature rather than boiling where the boiling point of the solvent controls the sample temperature. Heat is supplied by lamps; heated blocks/baths; or, in the latest generation of centrifugal systems, by low-temperature low-pressure steam. In these latest systems, water takes heat from the chamber wall, turns to vapor, then condenses on the sample vessels (which are cold due to the solvent boiling within) to transfer the heat to the samples. The heating vapor reaches all areas, minimal heat is wasted, and the time to reach the required temperature is very rapid, thereby improving the efficiency of concentration.
 Figure 6 – Rotary evaporation process.
Figure 6 – Rotary evaporation process.In addition to the effects of heating rate, the faster the vapors can be removed, the faster the solvents will boil. Since a wet, boiling sample is at the boiling temperature of the solvent, then the better a system is at getting heat into the sample, the faster concentration will occur, and the more efficient a system will be at removing vapors by condensation in a cold trap. However, although speed of concentration can increase with higher vacuum level, this remains true only to a certain point. At the highest vacuum levels achievable by modern evaporator systems, a volatile solvent will boil at extremely low temperatures that are so cold that a cold trap cannot effectively catch the solvent and is hence rendered useless.
For optimal system performance, a balance is therefore necessary between the heat input to the sample and the heat removal at the condenser. If a system is not balanced, either the cold trap does not capture solvent such that solvent goes to the pump, the pressure in the system rises, and solvent recovery is lost, or the cold trap effectively controls the pressure so that the system operates at a slower rate than optimal. In a freeze dryer, vapor flow rate is the controlling factor—the faster the vapor is removed, the faster the samples dry. This is most affected by cold trap power and temperature. Finally, concentration or evaporation can be accelerated by generating a greater solvent surface area, and blow-down systems speed up evaporation through a shift in equilibrium between phases.
Dr. Induka Abeysena is Application Specialist, and Rob Darrington is Product Manager, Genevac, The Sovereign Centre, Farthing Rd., Ipswich, Suffolk IP1 5AP, U.K.; tel.: +44 (0) 1473 240000; fax: +44 (0) 1473 461176; e-mail: [email protected].