Photostimulation techniques are among
the most powerful microscopy tools
available to life science researchers. They
are used in a variety of ways to illuminate
biological processes in action, allowing
the observation of fluorescent molecules
and the uncaging of compounds
that may then become biochemically
active. Such techniques as photoactivation,
photoconversion, and photoporation
are used to study living cells, tissues,
and organisms, typically using confocal
light microscopes. Photoconversion,
for
instance, is a vital tool for kaede studies
(Figure 1). Photobleaching,
the intentional
application of light at specific
wavelengths to cause photochemical
damage to a fluorophore, is often used
to examine motion or diffusion of molecules
when studying embryonic development
or doing calcium ratioing. The
observation of fluorescent molecules during
and after photobleaching using such
techniques as fluorescence recovery after
photobleaching (FRAP), fluorescence
loss in photobleaching (FLIP), and
fluorescence resonance energy transfer
(FRET) by acceptor photoactivation are
all widely used in laboratories throughout
the world. Seeing how cells recover
after photobleaching can yield clues as
to the extent of DNA damage; how and
when fusion proteins mature (green to
red); the behavior of photochromic proteins
as they increase or decrease fluorescence
or change color; the dynamics
of molecular mobility in the measurement
of diffusion, transport, and on/off
rates; the in vivo function of selectively
inactivated proteins of interest, as in
chromophore-assisted laser inactivation
studies in functional genomics; and the
processes of protein synthesis, embryonic
development, and stem cell activity
as observed through pulse labeling with
the addition of tags such as HaloTag
(Promega
Corp., Madison, WI) and
FlAsh and ReAsh (Invitrogen Corp.,
Carlsbad, CA).
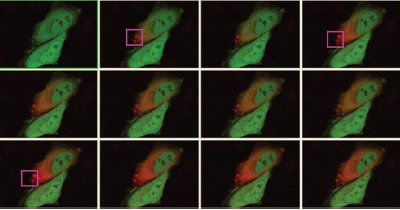
Figure 1 - Kaede photoconversion (green to red) using the Fluoview FV1000 confocal system with the simultaneous (SIM) scanner (Olympus America Inc., Center Valley, PA). (Image courtesy of Dr. Atsushi Miyawaki and Dr. Yasuko Ando, Brain Science Institute, Institute of Physical and Chemical Research [RIKEN] [Wako, Saitama, Japan].) (Image courtesy of Olympus Corp., Tokyo, Japan.)
Time lag and data loss
challenges
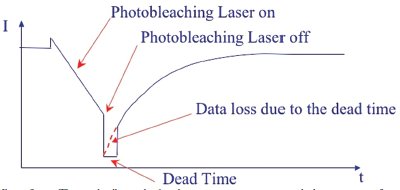
Figure 2 - The initial milliseconds after photoactivation ceases is a period where monitoring fluorescence recovery is not possible unless a second, simultaneous scanner is used. (Image courtesy of Olympus America Inc.)
Although photostimulation techniques
are very powerful, they do have several important limitations. First, there is a
significant lag time between the completion
of photoactivation and the start
of observation and documentation. In
a traditional FRAP or photoactivation
sequence, several reference images are
taken. The researcher defines a region
of interest within the reference image,
zooming in and increasing the power
of the stimulation source (laser in a
confocal system). The region of interest
is now exposed to this more powerful
excitation for a specified period
of time depending on the specimen
or experimental requirements. At the
completion of the FRAP or photoactivation
sequence, the system parameters
are reset to the original reference
image parameters to begin monitoring
for effects of the bleaching/activation
event. While the lag time for switching
back to normal imaging during a typical
FRAP experiment might last for milliseconds,
recovery starts as soon as the bleaching/activation event ends, leaving
a gap in the observation of highspeed
dynamic changes in the living
cells (Figure 2). In some experiments,
the most significant changes can occur
during the initial recovery “dead time.”
Scientists performing such experiments
are often forced to make assumptions
about what is happening during the
first milliseconds of recovery.
A related issue in photostimulation
experiments is that traditional scanning
methods have small gaps during
the scan process in which some
unmeasured recovery can occur. Specifically,
most confocal laser scanners
employ a raster scan technique, in
which the laser turns on, scans across
the specimen in a straight line, then
turns off, flies back across and down
to the start of the next line, turns on
again for the next sweep across, and
so on, until the entire region of interest is scanned. Though the process is
exceptionally fast, observation cannot
begin until the entire raster scan
is completed, and recovery starts to
occur intermittently as the laser turns
off during each fly-back.
In addition to issues with observation
time lag and unmeasured recovery,
conventional scanners incorporate
long photobleaching times, as intermittent
rounds of photobleaching and
recovery alternate until the fluorophores
lose their recovery potential.
Because of the necessity for long and
sometimes repeated exposures, experiments
incorporating FRAP and other
photoactivation methods face the
problems inherent in most traditional
confocal live cell imaging. These
issues include phototoxicity and photodamage.
The likelihood of eventual
cell death during the experiment
means that researchers often cannot
study the same sample repeatedly. In
addition, long-range time-lapse studies
become nearly impossible because
of the effects of long exposure to damaging
light.
Still another limitation of such experiments
is the difficulty in collecting
enough light from deep within specimens.
Although one of the strengths of
confocal microscope systems is in preventing
almost all out-of-focus light from
being collected, there is usually an inherent
loss of some light from the focal plane
as well, since some of this light is refracted
as it travels through the specimen. When
specimens are only dimly fluorescent, this
light loss can be significant. In addition,
signal-to-noise issues can arise when outof-
focus light is refracted into the collector.
Thus, refraction can be particularly
troublesome for observation occurring
deep within samples. This problem is further
exacerbated by the fact that light
from the specimen is detected far from
the objective in most confocal systems.
More signal is lost as the light travels
through scanning mirrors and the confocal
aperture before reaching the detector.
Solutions
Scientists compensate for the issues
of observation time lag, phototoxicphototoxicity,
and light loss in a variety of ways.
Software can offer a measure of help
with the dead time, since custom macros
built into a researcher’s software
can reduce the lost time and help
lessen the amount of data lost due to
unobserved recovery. Some researchers
have developed homegrown tools
to help them begin observation sooner
after photoactivation happens, but for
the most part, researchers know they
will lose some of this information and
design their experiments accordingly.
Issues with phototoxicity and collection
of insufficient light are pervasive
in confocal microscopy, and can
sometimes be addressed by using multiphoton
systems. Concerns about collecting
light from deep within specimens
can also be partially addressed
via multiphoton systems, since they
illuminate a single, focused point,
providing the system selected uses a
femtosecond pulsed laser and has its
detector near the objective.