One of the main focuses of the scientific community,
now that major advances have been achieved in the
comprehension of genome organization, has become
the study of proteins, including their structure and
activities. Among the different techniques available, mass spectrometry has played a fundamental role and
is experiencing rapid growth.1Matrix-assisted laser desorption ionization (MALDI) time-of-flight (TOF)/TOF instruments (Ultraflex Bruker
Daltonics, Bremen, Germany), taking advantage of
the reflectron mode, combine a soft ionization
process with good sensitivity and resolution.2
Until recently, many proteomic projects were aimed
at identifying proteins in biological samples in order
to characterize the proteomic profile of subcellular
compartments, cells, tissues, or pathological conditions.
A deeper comprehension of the protein
machinery will shed light on the complex and highly
dynamic protein network (interactome), how this
network transduces external stimuli inside the cell
(signalosome), and how it regulates cellular processes
(metabolome). Moreover, the identification of biomarkers
for severe pathological conditions, such as
cancer, heart disease, and neurodegenerative diseases,
will allow the establishment of early diagnosis and
the detection of pharmaceutical targets.
Experimentally, in peptide mass fingerprinting
(PMF),3 proteins, after enzymatic digestion, are
resolved in a number of peptides, whose masses are
determined and matched with a sequence database.
However, the presence of splicing variants in cells,
combined with the generation of different protein isoforms
or fusion proteins, gives rise to a complex picture
that demands a more detailed analysis.
Furthermore, the study of species with yet uncharacterized
genomes or the investigation of post-translational
modifications (PTMs) is not possible with classical
PMF, requiring simple, reliable de novo
sequencing. In the last 5–7 years, much effort has been
expended in order to improve the performance of peptide
sequencing with MALDI-TOF instruments.4
Described here is a fast, robust chemical modification
of peptides that strongly improves de novo sequencing,
using CAF™ (Chemical Assisted Fragmentation)
chemistry (GE Healthcare/Amersham Bioscience,
Uppsala, Sweden).
Principles and technical procedures
In MALDI instruments, postsource decay (PSD)5
produces nearly random fragmentation of the peptide
backbone, generating mainly b- and y-ions, but
other ions as well. In order to improve the peak
intensity and obtain easily interpretable spectra (i.e.,
only one series of ions), a chemical compound carrying
a sulfonation group is used.6–8 This group reacts
specifically with primary amines of peptide chains
(Figure 1b). In the first step (Figure 1a), ε-amino
groups of lysines are blocked, leaving the N-terminal
amino group the only one able to be sulfonated.
After reaction with the CAF reagent, a sulfo group is
transferred to the N-terminus. During the ionization
process, two protons are captured by
each peptide (Figure 1c). Postsource peptide
fragmentation generates b- and y-series fragments;
the y-series, carrying one proton, possesses
a single positive charge and will reach
the detector; however, the b-series, which carries
the negative charge of the sulfo group at
the N-terminus, after taking up the second
proton, becomes neutral and is therefore not
detected (Figure 1c). In this way, a single and
often complete series of y-ions is present in
the final spectra, leading to easy, safe interpretation
of the sequence.
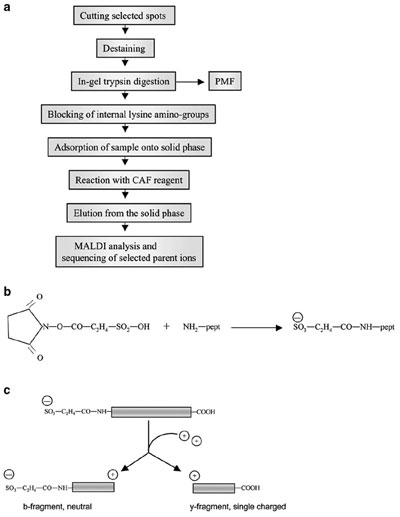
Figure 1 a) - Work flow of de novo sequencing with CAF chemistry. b)
Molecular formula of N-terminal sulfonation. c) Schematic representation
of charge distribution after peptide sulfonation. The sulfo group generates a
negative charge at the N-terminus. During the ionization and fragmentation
processes, two series of fragments, mainly b- and y-type, are produced, and
both capture a proton from the matrix. In the N-terminal fragment (bseries),
the positive charge of the proton is counterbalanced by the negative
charge of the sulfo group, generating neutral molecules that are unable to
reach the detector; only the C-terminal fragments (y-ions) carry a positive
charge and will travel in the mass spectrometer.
Proteins to be analyzed were separated by
polyacrylamide gel electrophoresis and stained
with appropriate methods. Selected spots were
cut out, destained as previously described,9
and incubated with trypsin. This generates
peptides with basic amino acids (lysine or
arginine) at the C-terminus, as required for
efficient fragmentation in the following steps.
The sulfonating reagent reacts with all primary
amino groups; in order to analyze lysine-ending
peptides, it is therefore necessary to
block the ε-amino group of lysines, leading to
peptides labeled with only one sulfonic group
(i.e., at the N-terminus). At least two methods
are available: 1) the addition of O-methylisourea
hydrogen sulfate (17.2 mg/mL
in 0.25 M NaHCO3, pH 10) and incubation
overnight at room temperature (RT), which
converts lysine into homoarginine in a guanidination
reaction (adding 42 Da/Lys), thus
eliminating the reactive ε-amino group, and
2) incubation of the peptide mixture with 2-methoxy-4,5-dihydro-1H-imidazole (Lys Tag
4H, Agilent Technologies, Palo Alto, CA) for
3 hr at 55 °C (adding 68 Da/Lys).
Peptide derivatizations were performed on solid-phase
supports: A μZipTip™ C18 (Millipore Corp.,
Bedford, MA) was wetted with a solution of trifluoroacetic
acid (TFA)/60% acetonitrile, then equilibrated
with 0.1% TFA. The peptide solution, deprived
of any organic solvent, was pipetted up and down
approx. 10 times in order to adsorb the peptides onto
the reversed-phase material. After column washing
with 0.1% TFA, the freshly prepared labeling solution
(1 mg/10 μL in 0.25 M NaHCO3, pH 9.4) was slowly
pipetted up and down, and allowed to react with the
adsorbed sample for 3 min at RT. The reagent was then
washed out with 0.1% TFA, and a solution of 5%
hydroxylamine in the same labeling buffer was pipetted
up and down a few times in order to remove unspecific
binding of the CAF reagent onto hydroxyl-containing
amino acid residues. The sulfonated peptides were
eluted by drawing 2–5 μL of 0.1% TFA/60% acetonitrile
up and down the tip. The sulfonation adds 136 Da
to primary amines in the peptide. A peptide with C-terminal
Arg will increase its mass by 136 Da, while
peptides with a C-terminal Lys will increase by 136 +
42 = 178 Da for guanidation Lys blocking, or 136 + 68
= 204 Da for imidazole incorporation (additional internal
Lys residues take up 42 or 68 Da each). If a Lys-containing
peptide was sulfonated without the blocking
step, it would be sulfonated also on the ε-amino group.
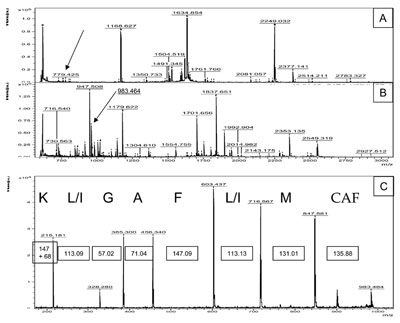
Figure
2 a) - PMF of a tryptic digest of cytochrome C from Candida krusei. b) The same peptide
mixture shown in (a) was Lys-blocked with imidazole (addition of 68 Da) and labeled at the N-terminus
with the sulfo group (addition of 136 Da). The MALDI spectrum after these modifications
shows more peaks and higher intensity compared to the native spectrum in (a). c) De novo
sequencing of the peak 983.464 (779.425 + 68 + 136). PSD of this peptide produces a neat series
of only y-ions. The bordered numbers show the difference in daltons between two adjacent peaks:
These masses fit with good precision with the theoretical masses of the corresponding amino acids.
For MALDI TOF/TOF analysis, samples were prepared
with the dried-droplet method: 0.3 μL of the sample
was mixed with an equal volume of a saturated solution
of ε-cyano-4-hydroxycinnamic acid (HCCA, Bruker
Daltonics, Bremen, Germany) in 0.1% TFA/40% acetonitrile;
a 0.3-μL drop was deposited on the polished
stainless steel MALDI target. All samples were analyzed
in reflector mode before and after derivatization to
obtain PMF spectra. By comparing these spectra and
scanning for additions of 136 or 178 (204) Da, candidates
for sequence analysis were identified. The instrument
was then switched to PSD mode and the ion
selector was set to the m/z values of the precursor ions
with a window ±0.2–1% of the parent ion mass.
Results
In order to test the efficiency of the CAF chemistry, the
authors used cytochrome C from Candida krusei. After
trypsin digestion and PMF, a number of peptides are
present in the spectrum (Figure 2a). The peptide mixture was then blocked at lysines with the imidazole
compound (addition of 68 Da per lysine), followed by
CAF modification. The presence of the imidazole
group renders the peptides more basic, thus enhancing
both the number of peptides detected and the intensity
of the peaks (Figure 2b). As an example, the authors
selected the peptide of 983.46 Da (see arrow in Figure
2b), arising from the diminutive peak of 779.42 Da in
Figure 2a (see arrow). PSD fragmentation of the peptide
gives rise to a clear series of only y-ions, generating
a clear and definitive amino acid sequence (Figure 2c).
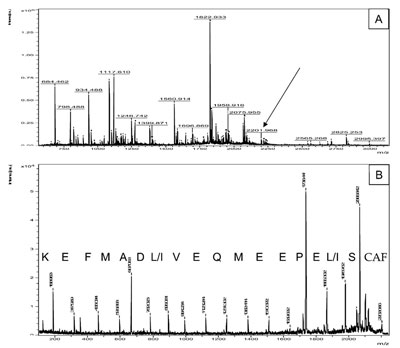
Figure 3 a) - Peptide mixture of an unknown protein from the mollusk Mytilus galloprovincialis
was Lys-blocked by guanidation, followed by sulfonation of the N-terminus. The figure shows
the MALDI spectrum of the modified peptides. b) PSD of the peptide of 2201.968 Da (see arrow
in [a]). The complete sequence of 17 amino acids is easily readable.
Although recent advances in the study of genomes have
made complete DNA sequencing of several species available,
the majority of them are still uncharacterized. For all
of these species, in the absence of a complete and reliable
genome database, identification of proteins with normal
PMF is impossible. Positive and unambiguous de novo
sequencing is a useful approach for proteomic studies of
this type of sample. In one proteomic study, the identification
of proteins from the bivalve mollusk Mytilus galloprovincialis
was required. After in-gel trypsin digestion,
matching the peptide list with databases did not provide
any protein identification due to lack of data for this
species (data not shown). The authors overcame this
problem by selecting representative peptides for de novo
sequencing. Peptides were first lysine-blocked with the
guanidination reaction, and then sulfonated on the N-termini.
Figure 3a shows the MALDI spectrum of one of
these sulfonated proteins. Figure 3b represents the case of
the peptide of 2002 Da (see arrow in [a]); it should be
noted that this peptide has a peak of quite low intensity
in the MALDI spectrum in comparison to other major
peaks. PSD fragmentation generated a clear sequence of
17 amino acids (SL/IEPEEMQEVI/LDAMFEK); a search
for homology with this sequence in the available database
permitted the identification of the protein as a cAMP-dependent
protein kinase. Hence, this approach allows
the identification of unknown proteins by sequence
homology, which is error-tolerant compared to PMF.
Conclusion
Increased proteomic interest has expanded the
demand for protein sequence information. To this
purpose, improved PSD after N-terminal sulfonation
provides a powerful and easy tool, suitable for
varied experimental purposes such as confirmation
of PMF protein identification, study of the proteome
from uncharacterized species, and analysis
of PTMs.
References
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature2003, 422(6928), 198–207.
- Stults, J.T. Matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS). Curr. Opin. Struct. Biol.1995, 5(5), 691–8.
- Thiede, B.; Hohenwarter, W.; Krah, A.; Mattow, J.; Schmid, M.; Schmidt, F.; Jungblut, P.R. Peptide mass fingerprinting. Methods2005, 35(3), 237–47.
- Standing, K.G. Peptide and protein de novo sequencing by mass spectrometry. Curr. Opin. Struct. Biol. 2003, 13(5), 595–601.
- Spengler, B. Post-source decay analysis in matrix-assisted laser desorption/ionization mass spectrometry of biomolecules. J. Mass Spec.1997, 32, 1019–36.
- Keough, T.; Lacey, M.P.; Youngquist, R.S. Derivatization procedures to facilitate de novo sequencing of lysine-terminated tryptic peptides using postsource decay matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2000, 14(24), 2348–56.
- Keough, T.; Lacey, M.P.; Youngquist, R.S. Solid-phase derivatization of tryptic peptides for rapid protein identification by matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom.2002, 16(11), 1003–15.
- Hellman, U.; Bhikhabhai, R. Easy amino acid sequencing of sulfonated peptides using post-source decay on a matrix-assisted laser desorption/ionization time-of-flight mass spectrometer equipped with a variable voltage reflector. Rapid Commun. Mass Spectrom. 2002, 16(19), 1851–9.
- Gharahdaghi, F.; Weinberg, C.R.; Meagher, D.A.; Imai, B.S.; Mische, S.M. Mass spectrometric identification of proteins from silver-stained polyacrylamide gel: a method for the removal of silver ions to enhance sensitivity. Electrophoresis1999, 20(3), 601–5.
The authors are with the Ludwig Institute for Cancer Research,
Box 595, SE-751 24 Uppsala, Sweden; tel.: +46 18 160423;
fax: +46 18 160420; e-mail: [email protected]. This
work was partly supported by fellowships to P. Conrotto from
Associazione Italiana per la Ricerca sul Cancro (AIRC). The
authors thank Dr. Antonio Villamarin, University of Santiago
de Compostela Lugo, Spain, for providing samples from the
mollusk Mytilus galloprovincialis.