In the development of many LC-MS
methods, interferences are commonplace.
In an environment in which data
must stand up to rigorous scrutiny, such as
in the pharmaceutical industry, selectivity
of an analytical method is very important.
Conditions required for achieving
this selectivity, whether they be related
to the sample preparation method, the
analytical column, or the MS detection,
dictate sample throughput.
Method selectivity is advanced by incorporating
FAIMS (high-Field Asymmetric
waveform Ion Mobility Spectrometry), a
technique that is orthogonal to and complementary
with both chromatography and mass spectrometry. This on-line gas-phase
ion separation delivers reduced chemical
background and separation of isobaric
endogenous interferences. This paper discusses
the new technology, which can be
easily implemented into LC-MS work
flows to increase laboratory productivity.
What is FAIMS?

Figure 1- Domed FAIMS electrodes (left) and side-toside
electrode (right).
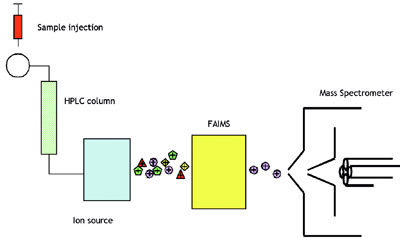
Figure 2- Schematic of HPLC-FAIMS-MS system.

Figure 3 -Domed version of FAIMS mounted on a Q-Tof
Micro MS (Waters/Micromass) (ion source, gas, and electrical
connections not installed).

Figure 4- Domed FAIMS mounted on a Q-Tof Ultima
MS (Waters/Micromass) (installation incomplete).
FAIMS is a technology that separates ions at atmospheric pressure. The FAIMS device is mounted between an atmospheric
pressure ion source and the entrance orifice to the vacuum chamber of the MS. Two examples of FAIMS electrodes are shown in Figure 1. A typical system incorporating FAIMS into an existing LC-MS system is shown schematically in Figure 2. Figures 3 and 4 illustrate
FAIMS mounted on two types of Waters/Micromass (Manchester, U.K.) mass spectrometers.
Three things are important about the technique: First, it separates ions in a manner quite differently from MS; second, it separates ions without loss of sensitivity (ion counts); and third, the ions flow continuously without the gates or drift times
associated with conventional ion mobility spectrometry. The simplicity, sensitivity, and selectivity via separation suggest that the technology will be of significant interest
to the LC-MS researcher. (A description of the mechanism of separation in FAIMS can be found at www.faims.com.)
FAIMS is compatible with condensed phase separations because it is physically
located between the source and the MS, and serves to sample the ion plume for the MS. As shown in Figure 2, an HPLC is
connected to an ion source, which may be
electrospray ionization (ESI) or atmospheric
pressure chemical ionization
(APCI). As the compounds are eluted
from the LC column they are ionized into
a flowing stream of ions. The stream of ions
is transported through FAIMS by a carrier
gas. Separation of the ions occurs while the
ions are carried along between a pair of
closely spaced FAIMS electrodes.
As shown in the figure, only
some selected ions from the mixture
are transmitted through
FAIMS. This flow of selected
ions is transferred to the MS.
Using software to control
FAIMS, the analyst selects the
ions that are carried through FAIMS by setting
two voltages, the dispersion voltage
(DV) and the compensation voltage (CV).
The dispersion voltage is the peak voltage
of a high-frequency asymmetric waveform
that is applied across the FAIMS electrodes.
The compensation voltage is a dc
bias between the electrodes that compensates
for the tendency of a given ion to drift
toward one of the electrodes. Fortuitously,
this CV is compound dependent. By selecting
the CV of transmission of the analyte,
other ions are removed by collision with
the FAIMS electrodes. Background and
interfering ions are therefore removed to
improve the signal-to-background ratio
(S/B) and potentially speed up analysis by
reducing the demands on the chromatographic
separation.
Quantitative analysis using
FAIMS
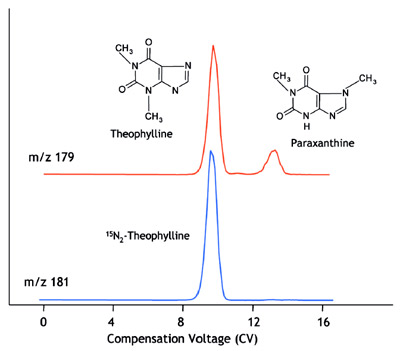
Figure 5 - Compensation voltage spectrum of a mixture of ions of theophylline and
paraxanthine acquired while monitoring m/z 179 (upper trace), and of isotopically
labeled theophylline by monitoring m/z 181 (lower trace). Traces are offset for clarity.
A chemical analysis begins with establishing
the practical values of DV and CV for
the target analyte compound. A standard
sample of the analyte is delivered by direct
infusion to the ion source. This gives the
user an opportunity to optimize operating
conditions of FAIMS (and perhaps the
MS). During this direct infusion step, the
DV is set to the maximum available on the
instrument, and the CV is scanned while monitoring the m/z of the target analyte to
produce a CV spectrum. The CV at which
the maximum signal is obtained is the CV
used for subsequent analyses. Unless experimental
conditions such as the temperature,
type of carrier gas in FAIMS, or gas
pressure in FAIMS are changed, the
FAIMS will be operated at this newly identified
CV for the duration of the analysis.
This is not unlike single ion monitoring
experiments using the MS.
Theophylline and paraxanthine, metabolites
of caffeine, are difficult to quantify
because they have the same m/z (and
therefore are not distinguished by MS)
and are difficult to separate using HPLC.
The traces in Figure 5 illustrate the CV
spectra taken during direct infusion-ESI
of two samples: The upper trace is a mixture
of theophylline and paraxanthine,
and the lower trace is a sample containing
only 15N-labeled theophylline.

After establishing the empirical value of
CV for the analyte (e.g., theophylline is
transmitted at a CV of 9.8 V, shown in Figure
5) the remainder of the analysis is performed
in the usual manner. The HPLC is
connected to the ionization
source, and samples are
injected. Standard samples,
including isotopically labeled
analogues of the target analyte and
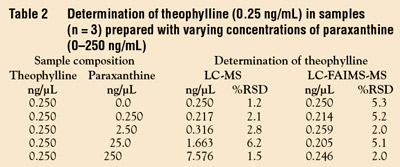
unknowns, are processed. Table 1 summarizes
experimental conditions for the LC-FAIMS-MS analysis, and Table 2 summarizes
quantitative measurements of samples
containing 0.25 ng/μL theophylline with
different added concentrations of the interfering
paraxanthine. The conventional
LC-MS results show anomalously high
results because paraxanthine coelutes, and
has the same mass as theophylline. Despite
coelution of the two compounds, the LC-FAIMS-MS results are quantitative.
Development of the procedure for
quantitation of theophylline in the
presence of paraxanthine using LC-FAIMS-MS required approx. a half-day.
HPLC runs were of ~5 min duration
without the need to separate theophylline
from paraxanthine. Calibration
for theophylline was linear over
the tested range from 0.1 to 10 ng/μL.
Separation of ions with identical
mass-to-charge ratio
Figure 5 illustrates a situation in which
FAIMS can be employed to separate
species that cannot be distinguished by
the MS and may also be difficult to separate
by HPLC. A quantitative determination
of theophylline is conducted at a
CV of 9.8 V, where the ion of theophylline
is selectively transmitted, as
shown in Figure 5. At this CV, any ions
of paraxanthine (having identical m/z to
theophylline) originating from the sample
will collide with the FAIMS electrodes
and not be introduced to the MS.
Similarly, paraxanthine is determined at
a CV of approx. 13.6 V without interference
by theophylline. Since these compounds
are not transmitted through
FAIMS under identical conditions,
development of the LC method is less
time-consuming because it is unnecessary
to separate these two compounds by LC.
Quantitative determination
of perchlorate
This example is selected to illustrate two
points. First, detection of ions of low m/z
with electrospray ionization is very difficult,
if not impossible, because of the
intense background ions. Second, perchlorate
is a sufficiently important analyte
from an environmental point of
view that a specialized analytical method
(U.S. EPA Method 314, www.epa.gov/safewater/methods/met314.pdf) involving ion chromatography has
emerged as the method of choice. It will
be shown that the analysis described
above for metabolites of caffeine can
readily be used for the detection of perchlorate
at levels appropriate for environmental
monitoring.