Fractionation is an essential step in the identification
of proteins and their subsequent structural
characterization. This information, in turn, is
vital to understanding protein participation and
function in normal and pathological cellular processes.
The vast range in protein abundance (at
least 6 orders of magnitude in dynamic range),
differences in solubility, and relative amenability
to separation by established methods (which
are often time consuming and manually intensive)
have created the need for new approaches
to fractionation that can overcome these limitations.
Specifically, problems that should be
addressed include:
- More efficient concentration, separation, and
capture of low-abundance proteins, which can
remain undetected in the presence of large-abundance
proteins
- Reduction in sample complexity by prefractionation
to increase the potential for detecting a
larger complement of proteins within a sample
- Separation of proteins that tend to be insoluble in
conventional gel separation buffers such as hydrophobic
and membrane proteins
- Concentration of fractions and minimization of
losses due to manual handling or other loss-prone
manipulations
- Reduction in fractionation processing time, which
greatly impacts R&D productivity.
One approach that has proven to be effective
in meeting these challenges is liquid phase or
free-flow isoelectric focusing (IEF). This technique
establishes a pH gradient within a liquid
buffer causing dissolved ampholytic molecules,
such as proteins, to migrate until they
reach a pH within the gradient corresponding
to their isoelectric point (pI)—i.e., they
have no net charge and therefore accumulate.
Such an approach has proven fruitful in concentrating
and/or purifying proteins with differing
pIs. Accordingly, IEF provides a means
for fractionating proteins that cannot be well
separated by size or affinity. Because it is an
orthogonal method, IEF can also be employed
as an adjunct to enhance separation and reduce
fraction complexity in conjunction with other
protein fractionation techniques.
In performing protein fractionation, it is necessary
to determine whether the sample has
been properly conditioned (qualification) and
is within the appropriate concentration range
(quantification). Quantification is important
in research planning, such as determining how
much of each fractionated protein analyte is
available for follow-up experiments. While satisfactory
quantification can be achieved with
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE), there are considerable
limitations associated with this method, in
particular, tedious and time-consuming manual
sample processing. For these reasons, it is highly
desirable to rapidly obtain relevant
analytical information so as not to
impede the pace of the work flow.
An elegant solution that meets
these requirements is an analytical
technique that utilizes electrophoretic-based separations within the
confines of a microfluidic chip. The
microscale dimensions of such chips
enable the rapid and accurate analysis
of very small sample aliquots of
either protein or RNA. Recently, two
instruments have been developed
that employ the two different adaptations
of electrophoresis indicated
for the respective fractionation and
analysis of microscale samples: the
MicroRotofor
™ system for protein
fractionation and the Experion™ system
for protein or RNA analysis (Bio-Rad Laboratories, Hercules, CA).
MicroRotofor
protein separation
system

Figure 1 - MicroRotofor system. The system is assembled according to the
protocol outlined in the instruction manual and filled with 2.5 mL of a properly
adjusted IEF ampholytic buffer (pH 3–10) containing 2.5 μg–2.5 mg of total
protein to be fractionated. The apparatus is then placed in the chassis prior to
separation. (Replacement of the focusing chamber is recommended after 4–5
runs with the same protein.) The system fractionates the sample into 10 discrete
200–250 μL fractions while applying continuous cooling in order to maintain protein
integrity. Sample temperature can be held at ~20 °C or ~10 °C, corresponding
to denaturing or nondenaturing conditions. After fractionation is complete,
the focusing chamber is moved to an internal harvesting station, containing 10
harvesting needles aligned to penetrate the focusing chamber at each fraction location.
Each fraction is drawn into its respective needle, and transferred by means
of vacuum aspiration, into the appropriate lane in the harvesting tray located
directly beneath the harvesting station cradle. The trays can be reused 5–10 times
and then discarded.
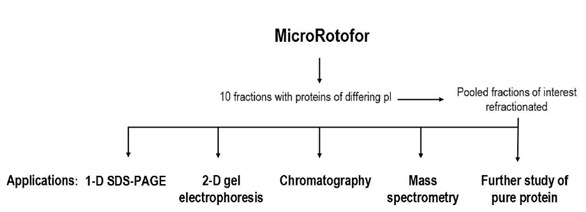
Figure 2 - MicroRotofor free-flow IEF—enhancement of protein fractionation.
A MicroRotofor separation is considered, in comparison with other protein
fractionation techniques, to be an orthogonal method, i.e., it employs a different
separation mechanism. More effective separations can generally be achieved with
two orthogonal methods employed in sequence than with any single technique
applied iteratively. The work flow depicted shows a variety of orthogonal methods
that can be enhanced by an initial MicroRotofor fractionation. In effect, the
10-band MicroRotofor fractionation becomes the first dimension of a second-dimension
or higher multidimensional separation scheme, with the downstream
dimensions selected to suit the specific application.
Advances in current tools for proteomics research have enhanced the
ability to work with smaller sample
volumes and protein amounts. The
MicroRotofor cell was designed to
meet this requirement. Like the
Rotofor® and MiniRotofor cells (Bio-Rad), the MicroRotofor cell utilizes
liquid-phase IEF, but on microscale
protein samples (~2.5 mL). In addition to reducing sample complexity
for follow-up fractionations and/or experimentation, MicroRotofor
IEF separations can be employed to
enrich the low-abundance component
of a protein sample, thereby
increasing the effective sample load
and/or dynamic range of detection.
The system’s high-resolution capability
facilitates the fractionation of
proteins that are typically difficult to
separate, such as closely related isoforms.
Separations can be run under
denaturing or nondenaturing conditions.
Figure 1 illustrates the system components
and outlines the IEF protocol. Figure 2
gives an overview of a number of separation
techniques that can be enhanced by coupling
with a MicroRotofor-based fractionation.
Applications
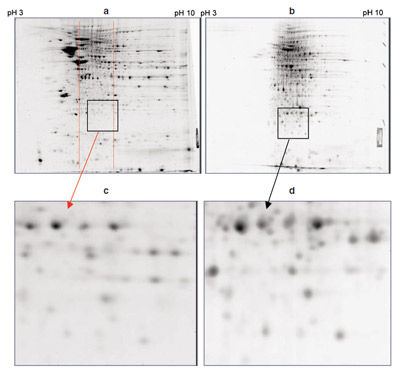
Figure 3- Enrichment of mouse brain proteins using the MicroRotofor cell. Comparison of 2-D gels of rat brain: a) unfractionated total protein (500 μg/gel), b) MicroRotofor fraction 5 (pH
7.5, 300 μg/gel), c) enlarged region (square) of unfractionated total protein gel in the same pH
range as the MicroRotofor fraction shown, and d) enlarged region (square) of MicroRotofor fraction
showing enrichment of many spots. Protein fractionation in the MicroRotofor cell improves
the 2-D resolution of low-abundance proteins that are not clearly detectable in the unfractionated
sample regardless of sample load. Increased sample loads of unfractionated sample simply lead to
increased streaking, which reduces resolution and obscures low-abundance proteins.
The utility of a MicroRotofor fractionation in combination
with other orthogonal separation mechanisms is demonstrated here.1 A proteomics application
work flow was modified by first using the
system on an E. coli lysate. Portions of the 10-band
MicroRotofor fractionation were harvested and
subjected to 1-D SDS-PAGE followed by in-gel
tryptic digestion of selected bands and LC-MS-MS analysis of the digest peptides. The results indicated
(data not shown) that each of the 10 fractions produced
10–50 protein bands in the follow-up SDS-PAGE
separation, and that 2–5 proteins were identified from each band. It is estimated that 500 or
more proteins could be identified by this approach,
which is comparable to the multidimensional protein
identification technology (MudPIT) approach
but is less complicated. Comparison with a conventional
2-D PAGE separation confirms that the multiplexed
MicroRotofor/SDS-PAGE approach gave
equal or better results in the number of proteins
detected. Moreover, the free-flow IEF fractionation
minimized sample loss due to insolubility problems;
eliminated the need to concentrate the crude protein
extract prior to separation; and enabled the
use of minigels for the SDS-PAGE step, simplifying
and accelerating the work flow upstream of the LC-MS-MS analysis.
In a separate enrichment experiment, two samples
of mouse brain protein were separated with
2-D gels. One sample was fractioned using the
MicroRotofor. Comparison of the pH 7.5 Micro-Rotofor fraction with the same pH region of the
unfractionated gel demonstrated enrichment of
spots attributable to low-abundance proteins in
the sample. This result was independent of sample
load (Figure 3).