High-speed calorimetry, and specifically
high-speed differential scanning calorimetry (DSC), has received a
great deal of attention in recent years.
Commercially, most calorimeters are
able to perform controlled, constant
heating experiments at rates higher
than a standard rate, typically 10
°C/min. Currently only one type of
calorimeter is available that is capable
of measuring at controlled, constant,
and appreciable high heating and
cooling rates: the power-compensation
DSC (PerkinElmer, Shelton,
CT, U.S.A.). Due to its small furnace,
strictly controlled cooling at, e.g., 300
°C/min and heating up to 500 °C/min
are possible, depending on the temperature
range of the measurements
and the cooling accessory used.
The reason high-speed calorimetry is
becoming increasingly popular is that
1) in practice, some processes occur at
much higher rates than realizable
using standard DSC; and 2) most
materials and substances, including
polymers and pharmaceuticals, are in
metastable states. Thermal history—
specifically cooling and heating
rates—and sample/product treatment
can change their behavior drastically,
including end properties. Such phenomena,
as related to metastability,
are well known to thermal analysts;
daily they encounter supercooling,
amorphization, cold crystallization,
recrystallization, annealing, etc.
Researchers need the capability to
change the measurement conditions in
order to influence and study the metastability
present. This article demonstrates
examples of metastability and the use of
high-speed DSC employing its commercial
versions: the Pyris 1 and Diamond
DSCs with the HyperDSC™ technique
(PerkinElmer).
Results and discussion
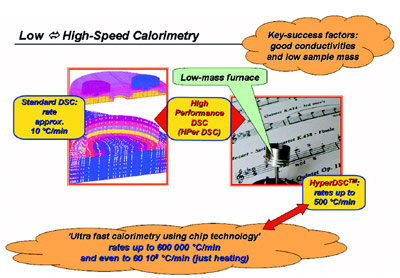
Figure 1 - Evolution of standard DSC toward a range of low- to high-speed calorimeters.
Because it is important in industry to
be able to drastically vary cooling and
heating rates, a project1 was initiated
in the late 1990s at DSM Research
(Geleen, The Netherlands) to address
this issue. It was realized that increasing
rates demand decreasing sizes of
relevant components of the measuring
equipment—especially the measuring
cell—in order to decrease the thermal
conductivity paths. Decreasing the
sample mass to values between 1 μg
and 1 mg is necessary also. Therefore, a
commercially available DSC was chosen
that offered the advantage of the
power compensation design, providing
direct heat flow rate measurements for
optimum results (see Figure 1). Of particular
significance are the influences
of the heating and cooling rate and
sample mass on temperature calibration
for high-speed DSC and specifically
calibration in cooling.1,2 This
approach was called High Performance
DSC (HPer DSC) and was later commercialized
by PerkinElmer and trademarked
with HyperDSC.
Figure 1 also depicts the ultrafast chip
calorimeters,3–5 which achieve extremely
high temperature rates (e.g., rates of
600,000 °C/min are possible), especially
in heating. However, they are not commercially
available, are not user-friendly,
and are time consuming to use. They are
well suited for scientific research purposes
but are not usable for daily practice
in industry.
Linking experiment with
practice and processing
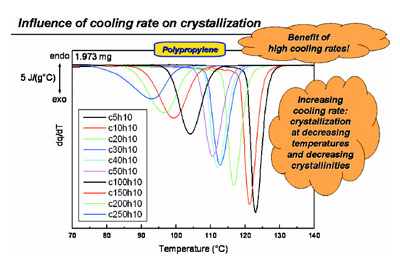
Figure 2 - Cooling of polypropylene at various rates and its influence on crystallization behavior.
As an example of an industry-relevant
experiment, in Figure 2 the drastic
reduction in the crystallization temperature
at increasing cooling rates is
shown for polypropylene. In addition,
the areas of the peaks are decreasing,
meaning that the crystallinity is lowered
as well.
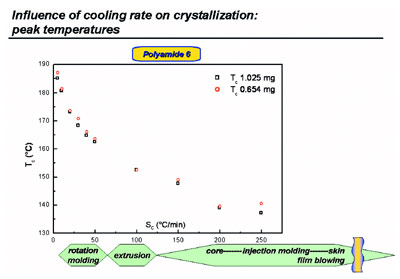
Figure 3 - Crystallization peak temperatures from cooling curves at various rates for polyamide 6.
Using the various cooling rates available,
some rates that occur during processing
can be mimicked (see Figure 3 for
polyamide 6). The cooling rate at the
skin of a mold is too high to mimic using
HyperDSC; therefore, a chip calorimeter
must be used.4,5 However, the exact
information on the actual cooling rates
as a function of the depth (from skin to
core) of a part processed is still missing,
because calculation by modeling requires
the right experiments, like the ones
shown here and in Refs. 1, 2, and 6.
In industry, this type of information is
critical because of the high expense of
the processing equipment used. Thus,
in processing, as in the case of injection
molding, optimization of the process—and specifically shortening of
cycle time—is of major importance.
Realistic information is also vital to
realizing fast iteration within the
“cycle of knowledge”7 in order to speed
up the materials development of new
grades and to optimize existing grades.
A striking example was presented in
Refs. 1 and 7. At 10 °C/min cooling—as
in the case of standard DSC—two peaks
were observed for crystallization of a
low-density polyethylene/linear low-density
polyethylene (LDPE/LLDPE)
blend. This would lead to the risk of segregation
by crystallization during film
blowing, resulting in numerous problems
in the film (i.e., optical and
mechanical). HyperDSC measurements
at 150 °C/min cooling show that it is
possible that no problem will occur at
all: Only one crystallization peak has
been observed. This must still be verified
for the even higher cooling rates in
the case of film blowing. Most likely,
extensive co-crystallization of molecules
of different origin takes place at this
(controlled) cooling rate. Obviously,
without the appropriate equipment, the
researcher/thermal analyst could easily
steer important and costly developments
in the wrong direction.
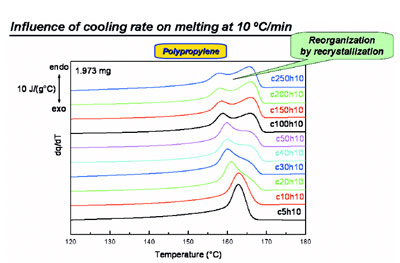
Figure 4 - Melting during heating at 10 °C/min after cooling at various rates for polypropylene
showing recrystallization behavior.
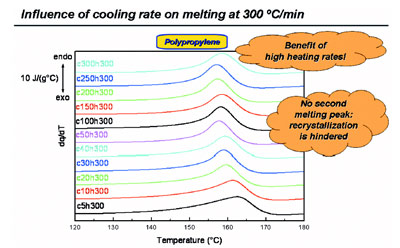
Figure 5 - Melting during heating at 300 °C/min after cooling at various rates for polypropylene
showing that recrystallization is effectively eliminated.
During heating at 10 °C/min (as in the
case of standard DSC) after cooling at
high rates, one often faces the problem
of extensive reorganization (Figure 4).
Two melting peaks are seen instead of
one following high cooling rates. Measurements
at varying heating rates
prove unequivocally that this reorganization
originates from recrystallization.
Heating at a higher rate of 300
°C/min (Figure 5) totally suppresses
recrystallization; in heating at this
rate, just one (the low-temperature)
melting peak results. Thus, recrystallization
is effectively eliminated.
In many instances, the relationship
between cooling (such as during injection
molding) and heating, and
between crystallization and melting, is
obscured by reorganization phenomena,
especially in the case of polymers and
pharmaceuticals.1,4,6 If this happens, the
relationship between the melting peak
temperatures and morphology information
obtained from measurements by,
e.g., X-ray transmission electron microscopy (TEM), and atomic force microscopy (AFM) performed at room
temperature is lost. Then, as a consequence,
important relationships
reported in the literature (such as the
Gibbs-Thomson relationship) no
longer apply. Therefore, many of the
reported studies would need to be
redone by applying much higher heating
rates to avoid such reorganization.
Quantitative measurements
Fast measurement takes minimal time,
and instrumental drift can be negligible.
Thus, it is possible to work quantitatively,
even at high rates, hence
the name High Performance DSC.
HyPer DSC provides more than high
speed. Ref. 1 gives examples in which
heat capacity measurements at rates
such as 100 °C/min are shown.
Recently,7 an extreme heating experiment
was presented on a 0.39-mg
high-density polyethylene (HDPE)
sample from –175 °C to 200 °C in one
run at 150 °C/min: a continuous heat
capacity measurement across a
tremendously wide temperature range
in just a few minutes.
Higher sensitivity; working
on minute amounts of
material
High rates increase sensitivity but
require small samples in order to
keep thermal lag acceptable. The
ability to obtain quantitative data on
small samples is an advantage also,
because it facilitates research on
minute amounts, which was previously
impossible.
How low can a sample mass be?
The authors learned that measurements
on samples down to 400 ng
(see Ref. 2) can be done. One
might ask: Is working with a few
micrograms still representative of
my product ? If the material is
homogeneous (modern materials
are produced more and more pure
while contamination during processing continues to decrease),
there is no problem. If not, the
contaminants, gels, discolored matter,
irregular surface parts, etc., can
be studied using HyperDSC.
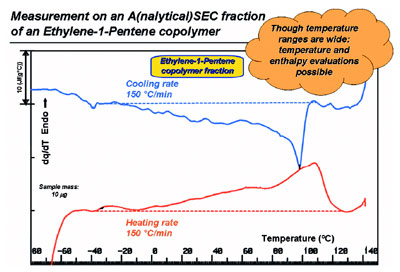
Figure 6 - HyperDSC on a molar mass fraction (Mn = 8–11 kgmol–1; Mw = 15–20 kgmol–1)
obtained by SEC from a whole, heterogeneous, metallocene-based ethylene-1-pentene copolymer
(Mn = 7.6 kgmol–1; Mw = 35 kgmol–1; 9.1 mol% of 1-pentene).
A good example of the use of the
low-sample mass capability of
HyperDSC is outlined in Figure 6.
Removal by evaporation of the eluent
used by a size exclusion chromatograph (SEC) provides a way to
deposit and spread the remaining
polymer sample on a germanium
disk as a function of molar mass: It
is fractionated according to molar
mass. It can then be measured by
FTIR to determine the short chain
branching content (if present) as a
function of molar mass. Removal of
fractions from the disk enables
measurement by HyperDSC. Since
the amount of starting material for
SEC (typically 800 μg of polymer)
has decreased over the years (20
years ago it was typically 5 mg),
HyperDSC is the only technique
capable of measuring (de)vitrification,
crystallization, melting, etc.,
in detail on such minute amounts.
The technique provides information on the distribution of the
short chain branches (SCB) as a
function of molar mass on the basis
of the crystallization and melting behavior, which then can be combined
with the average content
from FTIR.
Recently, the authors were able to
apply this method to a heterogeneous,
metallocene-based ethylene-1-pentene copolymer.8 This new
type of copolymer crystallizes and
melts across extremely wide temperature
ranges due to its broad ethylene
sequence length distribution;
still it can be measured effectively, as
is seen for the 10-μg fraction shown
in Figure 6. Obviously this can be
very useful, for instance, if only a
small amount of material is available
from polymerization.
Interesting and useful areas of
research are possible when only
minute amounts of material are available:
(sub)microscale synthesis, fractions,
explosives, nanostructures,
electronics, additives, contaminants,
multilayers, coatings, thin films,
skin-core problems in areas such as
chemicals, materials, products, and
forensic studies.
Degradation can be hindered or even
avoided by fast heating, in the same
way chemical changes during heating
can be prevented by simply spending
much less time in a critical temperature
range. The fast operation of
HyperDSC has been proven to enable
high-throughput experimentation.
Conclusion
Researchers who are willing to
explore high-speed calorimetry
instrumentation will become accustomed
to measuring at different
rates. As such, the intention of this
paper is not to promote the use of
high rates only, but rather to present
the option of choosing an optimal
rate or spectrum of rates,
depending on the sample and on the
question posed.7 This enables mimicking
real-life processes; and studying
the kinetics of processes of, e.g.,
polymers and pharmaceuticals,
including (hot, cold, re-) crystallization,
melting, reorganization,
annealing, de(vitrification), and
measuring on minute amounts of
substances, materials, and products
with high sensitivity in a short time.
As HPer DSC, commercially available
as HyperDSC, is increasingly
applied, more and more researchers
will learn how to benefit from this
exciting development.
References
- Pijpers, T.F.J.; Mathot, V.B.F.;
Goderis, B.; Scherrenberg, R.L.; van
der Vegte, E.W. High-speed calorimetry
for the study of the kinetics of
(de)vitrification, crystallization, and
melting of macromolecules. Macromolecules2002, 35, 3601–13.
- Vanden Poel, G.; Mathot, V.B.F. High
speed/high performance differential
scanning calorimetry (HPer DSC):
temperature calibration in the heating
and cooling mode and minimization
of thermal lag. Thermochim. Acta,
available on-line Mar 6, 2006, doi:
10.1016/j.tca.2006.02.022.
- Efremov, M.Y.; Warren, J.T.; Olson,
E.A.; Zhang, M.; Kwan, A.T.; Allen,
L.H. Thin-film differential scanning
calorimetry: a new probe for assignment
of the glass transition of ultrathin
polymer films. Macromolecules2002, 35(5), 1481–3.
- Tol, R.T.; Minakov, A.A.; Adamovsky,
S.A.; Mathot, V.B.F.; Schick, C.
Metastability of polymer crystallites
formed at low temperature studied by
ultra fast calorimetry: polyamide 6 confined
in sub-micrometer droplets vs.
bulk PA6. Polymer 2006, 47, 2172–8.
- Herwaarden, A.W. Overview of
calorimeter chips for various applications.
Thermochim. Acta 2005,
432(2), 192–201.
- Gabbott, P.; Clarke, P.; Mann, T.; Royall,
P.; Shergill, A. A high-sensitivity,
high-speed DSC technique: measurement
of amorphous lactose. Am. Lab.
2003, 35(16), 17–22.
- www.hyperdsc.com; see also webcast
by V. Mathot through this Web site; www.scite.nl.
- Luruli, N.; Pijpers, T.; Brüll, R.;
Grumel, V.; Pasch, H.; Mathot, V.B.F.
Submitted, J. Polym. Sci. Part B:
Polym. Phys.2007, 45, 2956–65.
Prof. Mathot is with SciTe B.V., Ridder
Vosstraat 6, 6162 AX Geleen, The Netherlands;
tel.: +31 6 14849332; fax: +31 84
8346102; e-mail: [email protected] and
with the Katholieke Universiteit Leuven, Heverlee,
Belgium. Dr. Vanden Poel is with DSM
Resolve, Geleen, The Netherlands. Mr. Pijpers
is with the Katholieke Universiteit Leuven.