Quantitative analysis of subvisible particles is an important unsolved problem, as discussed in the Particle Analysis track of PepTalk 2014, held January 12–17 at the Renaissance Hotel and Palm Springs Convention Center in California. Qualitative analysis is quite a bit better, but scientists still need better technology.
Protein aggregates are formed by intermolecular association of proteins or heteroaggregates with other materials, including particles. Aggregates are associated with altered potency plus potential immune response. Thus, drug developers and regulators are particularly interested in the qualitative and quantitative analysis of aggregates.
Aggregates are classed on the basis of size. Visible aggregates are usually larger than about 75 μm. Often these show up as filamentous particles or cloudiness of the liquid as it is prepared for administration. Patients and providers respond to this by questioning the suitability for injection. However, it is the subvisible aggregates that are of the most concern, since one needs expensive equipment to confirm their presence. Even if they are present, there is no good technology for measuring their concentration. Hence, there is little information on safe concentrations.
Proteins by their nature have evolved to interact with other molecules in living systems including DNA, RNA, carbohydrates, hormones, and other proteins. For protein drugs, therapeutic doses are often in the 100-mg range. For administration, many of these are dissolved or suspended in 1 mL of aqueous buffer. This is a very concentrated solution (~10% solids). Worse, the packaging process involves lyophilization in the vial. A small aliquot of the drug formulation is placed in the vial and then dried in place. This means that the concentration gets very high as the last liquid is removed. The laws of mass action operate to force the proteins together, usually forming aggregates. Over time, even in the dry state, the proteins may continue to react with their close neighbors. This explains the formation of covalent and hence irreversible aggregates.
A further complication is that aggregate properties responsible for immune response are not understood. For example, the adjuvant effect in vaccines is still mostly empirical (see en.wikipedia.org/wiki/Immunologic_ adjuvant). Protein aggregates form dynamically, usually according to the laws of mass action. Others may covalently cross-link. Another problem is that the physical form of the aggregates is not uniform so that size is often not related to concentration. Thus, one needs to be careful to specify the units for quantitative analysis of a particular size range, since the particles may have different structures, weights, and densities. Plus the volume and mass scales with the radius cubed at constant density.
Table 1 – Summary of techniques for measuring protein aggregates discussed at PepTalk 2014*
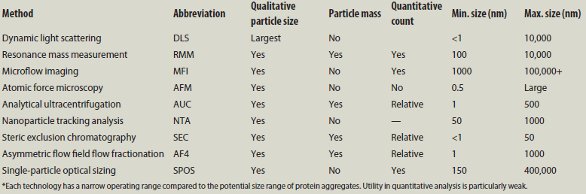
Several lectures described methods that are used for qualitative information about the aggregates. Prof. Elizabeth Topp of Purdue University (Lafayette, IN) summarized the first session, as shown in Table 1.
Dynamic light scattering (DLS) is one of the oldest technologies for measuring particle size. DLS responds selectively to the largest particles in the detection cell, so one often uses it in conjunction with a separation unit such as steric exclusion chromatography (SEC) or asymmetric flow field flow fractionation (AF4).
Resonance mass measurement
Resonance mass measurement (RMM) utilizes a flow channel in a resonating cantilever. When a particle is in the channel, the mass changes, which changes the resonance frequency of the cantilever. The solution must be dilute so that the probability of having two or more particles simultaneously in the channel is reduced. Since many protein formulations are very concentrated, RMM has not been generally successful in providing useful information.
Microflow imaging
Microflow imaging (MFI) uses flow microscopy to image and count particles. It is beneficial for particles larger than about 1 μm, and certainly larger than 0.1 μm. The particles need to be dilute enough to see individual images.
Atomic force microscopy provides high-resolution images of molecules and particles on a surface. Thus, AFM is used for qualitative analysis of individual particles, but throughput is slow.
Analytical ultracentrifugation (AUC) is a well-established technique for separating particles based on their sedimentation velocity. Throughput is a run/day at best, and discriminating power is modest.
Nanoparticle tracking analysis (NTA) uses an ultramicroscope with laser illumination. A charge-coupled device (CCD) camera records the Brownian motion of individual particles, which is governed by viscosity and temperature. Software provides the hydrodynamic radius distribution of the suspension in the range of 10–1000 nm. Since individual particles are counted, NTA avoids the problem of DLS selectively responding to the largest in the population.
Steric exclusion chromatography
Steric exclusion chromatography (SEC) is often thought of as a possible technology for qualitative and quantitative analysis. However, SEC requires a dilute solution. Dynamic aggregates can disaggregate at low concentration. Analytes can be adsorbed by the column packing or be shared by the flow. Thus SEC is usually limited to analytes smaller than a few megaDaltons.
Asymmetric flow field flow fractionation
Asymmetric field flow fractionation (AF4) separates molecules based upon differential diffusion coefficients in free solution. If the particles have similar density, the particles are separated by size. AF4 is often used with multiangle light scattering to further characterize the particles emerging from the separation chamber or hollow fiber. AF4 is not considered reliable in quantitative analysis.
Single-particle optical sizing
Single-particle optical sizing (SPOS) from Particle Sizing Systems, LLC (Goleta, CA) illuminates a colloidal suspension with a laser and measures light extinction for particles 0.5 μm and larger. Light scattering is used for particles in the range of 150–500 nm.
As an example of the idiosyncracies involved in measuring particles, Prof. Topp compared the results from DLS and RMM as a function of concentration of 1000-nm polystyrene standard particles. At 1010 particles/mL, the response indicated a dp of 20 nm. At 108–107 particles/mL, the results were about right, but at 106 the size fell off to 500 nm. In contrast, RMM provided correct results for 400-nm standards over the concentration range of 109–107 particles/mL. With RMM, there is a practical lower limit of acceptable measurement time for dilute suspensions.
Next, Prof. Topp asked what processing steps make a difference. Using TNF-α as an immunogenic indicator, and thermally stressed IgG as the agonist, she found that agitation and stirring at pH 3 increased production of TNF-α. The last point in her slide deck understated the current status: “Aggregate properties responsible for immune response are not well understood.”
Thus, the details of protein aggregation need study. Prof. Tom Laue (University of New Hampshire, Durham) provided an overview in a plenary lecture entitled, “Mechanisms of Protein Aggregation.” This focused on the crowding effect of concentrated solutions as is often found in antibody therapeutics. As one dries a concentrated aqueous solution, water protecting hydrophobic patches on the proteins is removed. This lowers the activation energy barrier for aggregation and possibly other reactions, including deamidation.
Prof. Laue compared several molecule–molecule interactions including charge–charge. He concluded that charge–charge repulsion is the safest way of avoiding aggregation. However, he cautioned that the charges must be measured and not just calculated, since huge differences are common, especially in complex and concentrated solutions.
Mitigating aggregation
The bulk of the program presented case histories addressing what can be done to avoid aggregation. The short response was:
- Modify the protein to increase charge
- Modify bioprocessing conditions to avoid process steps and conditions favorable to aggregation
- Store protein products in dilute acids.
Two lectures focused on hydrogen-deuterium exchange as a method to study regions of stability for protein structure. In general, the more accessible an amide proton, the more rapid the replacement of the proton by deuterium, which shows up as an increase in MS mass by 1 amu. Time frames for exchange range from 101 to 105 sec.
Dr. Prakash Manikwar of Medimmune (Gaithersburg, MD) lectured on improving the stability of protein therapeutics with excipients. Excipients can improve stability apparently by shielding potentially reactive groups from each other. Differential scanning calorimetry and steric exclusion chromatography are also effective in improving stability.
Dr. Jeffrey Hudgens of NIST used hydrogen-deuterium exchange to study protein–protein interactions. One cited example was that an unstressed α-helix is very slow to exchange, but when extended (stressed), the exchange rate increases dramatically. NIST’s HDX-MS system starts with a multiarm process robot for sample preparation, including protein digestion. An ultrahigh-performance liquid chromatograph (UHPLC) running at 1 ºC is connected directly to an Ion Trap/Orbitrap MS (Thermo Fisher Scientific, Waltham, MA). This process is capable of precision of 2%.
The specific example from Dr. Hudgens was proliferating cell nuclear antigen (PCNA), which is a trimer that circles duplex DNA and binds to DNA polymerase to increase processing. PCNA has many other partners involved in DNA transcription and repair. As expected, deuterium update is more rapid in the outer loops of PCNA. The team used site-directed mutagenesis to change amino acids at various positions to elucidate the nature of the segments responsible for association and synthesis activity. Also, as a work in progress, the NIST team is looking for advanced software that will help evaluate complex interactions of multiple protein ensembles
Dr. Vishal Nashine and colleagues at Bristol-Myers Squibb (New York, NY) report that methionine oxidation is a common degradation pathway for proteins. Two potential mechanisms exist. The one-electron pathway involving free radicals is stimulated by metals and light. Surfactants and polymers facilitate the two-electron pathway. The team is developing models to help explain and differentiate between the mechanisms of action.
A lecture by Dr. Jerry Lampert (Eli Lilly & Co., Indianapolis, IN) reported using a conventional design-of-experiments approach to find conditions that avoid aggregation of a large peptide product. They developed a simple test to remove aggregates with a 100-kD ultrafiltration membrane and UV photometry to measure the amount of peptide lost. Reducing the pH to 20º from higher storage conditions was effective in reversing aggregation and stabilizing the product.
Aggregation of drug-antibody conjugates (ADC) was the topic of a lecture by Dr. Dimiter Dimitrov of the NIH (Bethesda, MD). A large fraction of the anticancer therapeutics in development are ADCs using antibody fragments for immune targeting. He focused on CH2 domains since these are generally conserved, which might increase the therapeutic range, but CH2 fragments have a reputation for being prone to aggregation. Using sitedirected mutagenesis, he was able to reduce aggregation by incorporating a new –S–S– bond between the A&G units. While this avoided aggregation, adding the drug conjugate increased aggregation. This is a work in progress. Dr. Dimitrov is hopeful that new models and protein engineering will be successful in reducing or eliminating aggregation for these ADCs.
Conclusion
PepTalk 2014 was organized by Cambridge Healthtech Institute (CHI) in Cambridge, MA. I was impressed with the high-quality program that CHI organized on this important and complex topic. Protein aggregation suffers from lack of good analytics, especially covering the range of about 100 kDa (~1 nm) to 1 μm. This is a 103 range in size and a 109 range in mass. The speakers pointed out the problem and described the best available tools. From their reports it is evident that we need better ones.
Addendum
Since the writing of this article, two things popped up: The first was a high-resolution image1 obtained by crystallography of six IgGs forming a hexagon complex that activates the compliment. This explains how circulating antibodies can associate via biospecific, noncovalent interaction of the Fc regions of the antibodies, which triggers the compliment cascade, leading to destruction of target cells.
Since a majority of the drugs in development are based upon antibodies, including drug antibody conjugates, one should not be surprised that antibodies agglomerate. Aggregation is precisely what they evolved to do to protect us. This may also be relevant to the adjuvant effect where adding a sorptive agent (typically alum) increases potency in vaccines. The agent may sorb the antibodies, which favors formation of the hexamer.
The second advance is a novel Aggregation Analysis System for Biopharmaceuticals from Shimadzu Scientific Instruments (Columbia MD).2 This instrument uses diffraction from a violet laser to estimate the size of subvisible aggregates 50 nm or larger. Output is either cumulative or differential with bimodal capability. Polystyrene latex particles are used for reference. Concentration range is low ppm. This limits utility in concentrated solutions often found in antibody formulations that are often in the percent range.
Reported applications include aggregation of gamma globulin in various buffers, and growth of aggregates in 15 hr with 1-sec resolution. Another studied the effect of stir time on aggregation. One example showed suppression of bovine serum albumin (BSA) aggregation by adding L-arginine to the liquid.
References
- Anon. Hexing Complement. Science Mar 2014, 343, 1175; Diebolder, C.A. et. al. Science Mar 2014, 343, 1260‒3.
- http://www.ssi.shimadzu.com/products/product.cfm?product=aggregates-sizer_2
Robert L. Stevenson, Ph.D., is Editor, American Laboratory/Labcompare; e-mail: [email protected] .