The classical example of detrimental effects caused by bacteria, such as E. coli or Bacillus Anthracis, is through invasion of the host. Additional toxins are another mechanism that exists for harmful or potentially lethal effects from bacteria or fungi. Referred to as toxigenic fungi, the more widely studied of the sefungi include Aspergillus, Fusarium, Penicillium, Stachybotrys, and Mycothecium. In many instances, these fungi do not even need to be actively growing, but were merely once present under the correct growing conditions for the toxins to have been produced and contaminate the food source. Importantly, these toxins are not required for fungal growth and are simply a product of the primary metabolite process of the fungi (a secondary metabolite).
These secondary metabolites of fungi are referred to as mycotoxins and their function is not completely understood. It is believed that they are produced by the fungi to eliminate organisms that might be competing for the same resources in their growth environment. It has been found that a single mycotoxin can cause more than one type of effect; some are carcinogenic, some are vasoactive; others affect the central nervous system. As an example of the potential toxicity of these toxins, ochratoxin (produced by A.ochraceus) has been found to have an LD50 (lethal dose for 50% of a population) of approximately 22 mg/kg body weight.1
Because many of these fungi frequently infect economically important plants, the cost of a large contamination can be substantial. Mycotoxin contamination is recognized as an unavoidable risk because the formation of fungal toxins is weather dependent, and effective prevention is difficult. According to the Food and Agriculture Organization (FAO), more than 25% of the world’s agricultural production is contaminated with mycotoxins, leading to economic losses of $923 million annually in the United States grain industry alone.2
The health hazards, potential economic loss, and unavoidable risk of contamination demand sensitive, robust, and specific analytical methodologies. Current methods include gas chromatography with electron capture3 or mass spectrometric4 detection, liquid chromatography with postcolumn derivatization and fluorescence detection,5 and liquid chromatography with tandem mass spectrometry (LC-MS-MS).6
LC-MS-MS appears to have the most promise as a highly specific, broadly applicable detection scheme that provides both qualitative and quantitative data. Current LC-MS-MS methods, however, are unable to detect all the relevant groups of mycotoxins in a single run and cannot meet many of the stringent ion ratio reproducibility requirements of some European Union directives.
Experimental
All mycotoxins, except fumonisin B3, were purchased from Sigma-Aldrich (St. Louis, MO). Fumonisin B3 was a generous gift of Darsa Siantar, Alcohol and Tobacco, Tax and Trade Bureau (TTB, Walnut Creek, CA). All LC solvents were purchased from Fisher Scientific (Boston, MA) as HPLC grade or better, and all other reagents were purchased from Sigma-Aldrich.
Table 1 - Gradient program and LC/API conditions
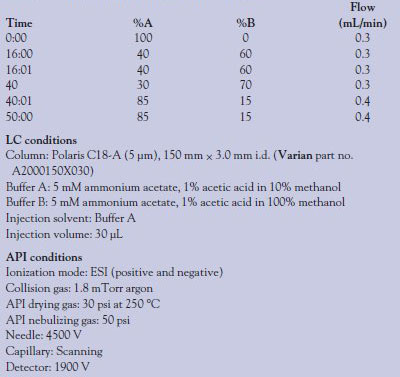
All equipment, unless noted, is from Varian, Inc. (Palo Alto, CA). The LC-MS-MS system utilized in these experiments is the 320-MS triple quadrupole mass spectrometer fitted with an electrospray ionization (ESI) source, two Pro-Star™ 210 liquid chromatography pumps, and a ProStar 430 autosampler. Separation was according to the gradient program shown in Table 1. LC conditions and mass spectrometer (atmospheric pressure ionization [API]) conditions are also shown.
Fully automated solid-phase extraction (SPE) and sample preparation were performed on a Zymark SPE workstation (Hopkinton, MA) according to the following method: Prior to extraction by the SPE system, sake wine or wheat beer was degassed by sonication for 30 min at room temperature. The degassed wine/beer was filtered through Whatman no. 1 filter paper (Florham Park, NJ), and the solution of multiple mycotoxins was added to a final concentration of either 35 or 350 ng/g (see text) with an internal standard (ZAN) concentration of 50 ng/g. This mixture was applied to a Bond Elut® mycotoxin SPE column (part no. 12165001B) without preconditioning the column. Samples were eluted with methanol and 30 μL was injected into the LC-MS-MS (Table 2).
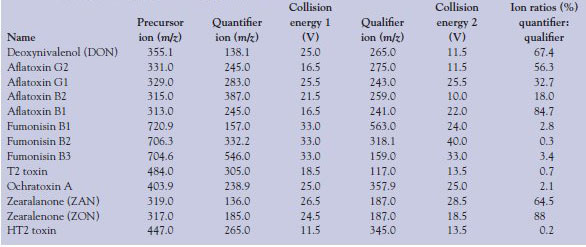
Table 2 - MS-MS conditions for each mycotoxin including starting mass (precursor ion), the collision energies used to fragment the precursor ion, and their respective qualifier and quantifier product ion; the ion ratio percentage (quantifier to qualifier) is also given
Results and discussion
The most widely used method for the detection of mycotoxins is HPLC separation with direct, postcolumn derivatization and UV-VIS or fluorescence detection. The disadvantage of this method is twofold: 1) It requires a separate analysis method to obtain the optimal resolution necessary for accurate quantification, and 2) it does not provide any means to positively identify the mycotoxin (confirmation analysis). By contrast, the LC-MS-MS method presented is rich in quantitative and qualitative information and has the accuracy and resolution required for reporting results to European and U.S. governmental agencies.
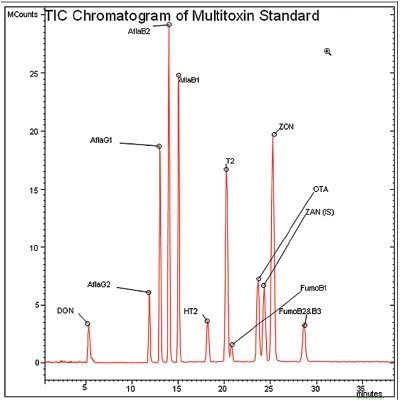
Figure 1 - Detection of 12 mycotoxins by LC-MS-MS. Peaks shown are deoxynivalenol (DON), aflatoxin G2 (AflaG2), aflatoxin G1 (AflaG1), aflatoxin B2 (AflaB2), aflatoxin B1 (AflaB1), HT2 toxin (HT2), T2 toxin (T2), fumonisin B1 (FumoB1), ochratoxin A (OTA), zearalanone (ZAN) (internal standard), zearalenone (ZON), and fumonisin B2 and B3 (FumoB2 and B3). Concentrations for DON; ZON; T2; HT2; and fumonisins B1, B2, and B3 are all 400 ppb. OTA is 170 ppb and all aflatoxins are 40 ppb (on column).
Table 3 - Calibration curve range, linearity (r2), LOD, and suggested maximum levels for each mycotoxin
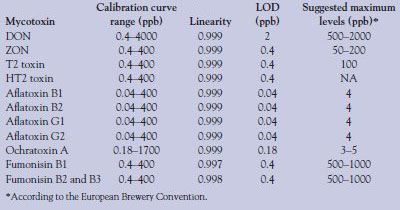
Figure 1 shows the separation and detection of all relevant mycotoxins in a 35-min analytical run with a 30-μL injection. Excellent separation is obtained for all the mycotoxins tested, with the exception of fumonisin B1 (21 min). Individual mycotoxins were injected, in series, at very low levels to determine the limit of detection (LOD) of each mycotoxin. Calibration standards for individual mycotoxins were prepared (starting at the LOD) in injection buffer (Buffer A conditions) by adding a known concentration of each mycotoxin to individual tubes and serially diluting the samples with Buffer A. Calibration ranges were different for individual mycotoxins and are given in Table 3. These samples were injected and analyzed, and 4–5 point calibration curves (three replicates per point) were determined. Table 3 shows the results for each mycotoxin including LOD. Good linearity and reproducibility are obtained with this method down to the parts per billion range for each of the mycotoxins, with only one mycotoxin showing a greater than 0.5-ppb detection limit. In some cases, the LOD of the individual mycotoxin is as low as 40 ppt, an extremely low level. Table 3 also shows the limits of detection required by the European Brewery Convention for beer in the European Union. Detection limits for this method exceed the required limits for all the mycotoxins that require testing, by many orders of magnitude in some cases.
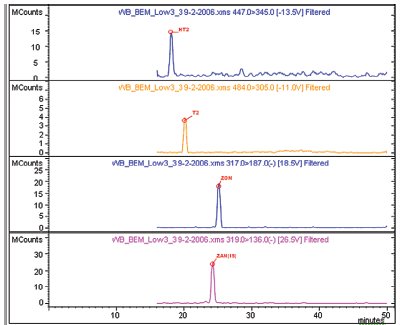
Figure 2 - Example chromatograms of low-level spikes for various mycotoxins in a wheat beer matrix. Shown (top to bottom) are: HT2 toxin (HT2), T2 toxin (T2), zearalenone (ZON), and zearalanone (ZAN) (internal standard).
To test the potential of this method in a real-world matrix, a mixture of mycotoxins was spiked into a complex matrix of wheat beer and/or sake wine. After the initial spiking of the beer or sake, sample cleanup was performed by SPE of the sample (see Experimental section). Figure 2 shows example chromatograms of low-level (35 ng/g) spikes of several mycotoxins in a wheat beer matrix. Shown are HT2, T2, ZON, and ZAN (internal standard), and all have excellent peak shape and signal in each of the runs. Because a known concentration of mycotoxin is spiked into the matrix, recovery rates of the toxins can be calculated to validate the SPE method. Table 4 shows the recovery rates (% recovery) of several tricothecene family members from wheat beer and sake. With the exception of T2 toxin (high and low concentration) and the low concentration of HT2, rates of recovery are excellent; all are greater than 80% with very good reproducibility (n = 3).
Table 4 - Rates of recovery for selected mycotoxins by solid-phase extraction from wheat beer and sake at two concentrations; relative standard deviation is calculated from three replicates
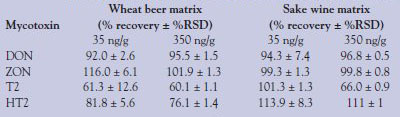
It is not known why the T2 toxin is difficult to recover. Further investigations into optimizing the extraction procedure are necessary. Similar recoveries of T2 toxin, DON, ZON, and HT2 from cereal and millet matrices were reported by Kloetzel et al.7 using the same Bond Elut mycotoxin cartridge. In EU guideline 2005/38/EG, the recovery limits are given as follows: 60–120% for DON/ZON and 60–130% for T2/HT2. The recovery rates of the newly developed method are in these ranges and, thus, the method satisfies the EU requirements. Also of note is that recoveries appear to be lower in wheat beer than in sake. It is possible that a component of the wheat beer matrix is interfering with the recovery, or the column is simply overloaded by some aspect of the matrix and an ion-suppressing factor from the matrix is passing through, inhibiting the detection of the T2 ion in the mass spectrometer.
Conclusion
A rapid and reliable method for the separation and detection of mycotoxins from a complex matrix is described. The Bond Elut SPE cartridges provide a fast, simple, and inexpensive ($3/cartridge) way to isolate the mycotoxins from wheat beer or sake, and liquid chromatography tandem mass spectrometry offers an ideal detection method. LC-MS-MS provides both qualitative and quantitative information and allows for the accurate identification of the mycotoxin, which is nearly impossible for fluorescence methods to accomplish. The method also fulfills the required limits of detection as well as the ion ratio requirements for European Directives such as SANCO D.1(06)D/412820.
References
- Braun, M.H.; Szczech, G.M.; Pumalis, B.P. Toxicol. Appl. Pharmacol.1976, 37(2), 331–8.
- World Wide Regulations for Mycotoxins in Food and Feed in 2003. FAO Food and Nutrit. 2003, 81, 3.1–3.5.
- Scott, P.M. J.Assoc. Off. Anal. Chem. 1986, 65, 876.
- Milanez, T.V.; Valente-Soaresh, L.M. J. Braz. Chem. Soc. 2006, 17(2), 412–16.
- Thiel, P.G.; Stockenstrom, S.; Gathercole, P.S. J.Liq. Chromatogr.1986, 9(1), 103–12.
- Biselli, S.; Hartiq, L.; Weqner, H.; Hummer, C. LC·GCEur.2004, 25–31.
- Kloetzel, M.; Lauber, U.; Hump, H.U. Mol. Nutr. Food Res. 2006, 50, 261–9.
The authors are with Varian, Inc., 2700 Mitchell Dr., Walnut Creek, CA 94598, U.S.A.; tel.: 925-939-2400; fax: 925-945- 2206; e-mail: [email protected].