Genome sequences and proteomic data are becoming available to researchers for an increasing number of normal genes and different pathologies. Characterization of these genomes and proteomes will be critical for the proper evaluation and treatment of disease. DNA microarrays, chemical genomics (proteomics), pharmacological genomics (proteomics), and bioinformatics can be used to achieve these goals. This article discusses how traditional and novel chemical and pharmacological techniques for genomics and proteomics can be used to treat disease and enhance drug discovery and development.
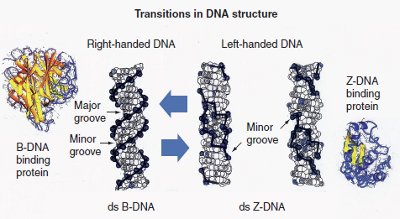
Figure 1 - Potential B-DNA to Z-DNA transition that occurs in some ds-DNA sequences. This process can be regulated by DNA binding proteins. Right-handed double-stranded (ds) B-DNA has a smooth backbone with major and minor grooves. On the other hand, left-handed ds-Z-DNA has only a minor groove. This figure represents structural transitions that occur in conventional ds-DNA and result in alternative structures. (Reproduced with permission from Future Medicine Ltd. [London, U.K.])
Four novel genomic and proteomic techniques have been developed that are the first practical applications of the novel multistranded and alternative DNA, RNA, and plasmid microarrays (transitional structural nucleic acid microarrays):1–3 transitional structural chemogenomics (TSCg), transitional structural chemoproteomics (TSCp), transitional structural pharmacogenomics (TSPg), and transitional structural pharmacoproteomics (TSPp) (Figure 1).4–6 These next-generation genomic and proteomic technologies will help in the study of conventional, alternative, and multistranded DNA, RNA, DNA–protein complexes, RNA–protein complexes, nucleic acid–protein–protein interactions, and nucleic acid– protein–drug interactions.
The microarrays shown in Figure 21–3 can be used together with conventional chemogenomics, chemoproteomics, pharmacogenomics, and pharmacoproteomics, and TSCg, TSCp, TSPg, and TSPp4–6 to improve the characterization of DNA and RNA structure, gene expression, nucleic acid–protein interactions, nucleic acid–ligand interactions, drug discovery and development, and nanomedicine.
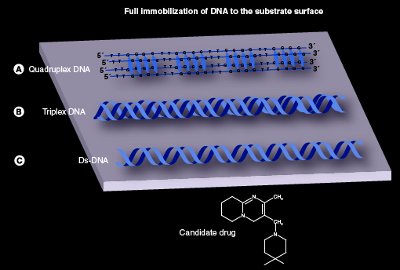
Figure 2 - This figure reveals how three different nucleic acid molecules can be immobilized to a solid support, viz., substrate surface: a) quadruplex DNA, b) triplex DNA, and c) ds-DNA. The three different nucleic acids can be tested for their ability to bind with drugs and/or proteins under different environmental conditions. Quadruplex DNA and triplex DNA are examples of multistranded DNA molecules. The ds-DNA represents conventional DNA.
Novel technologies for gene expression studies and drug discovery and development
TSCg, TSCp, TSPg, and TSPp (Figure 3)2–6 remove many of the obstacles associated with gene expression studies and drug discovery and development. Traditional DNA microarrays have mainly been used to identify changes in gene expression.7–13 They are based on the primary structure of DNA molecules, that is to say, the base sequence of single-stranded (ss)-DNA (i.e., DNA probe) that will hybridize with a target element. The end result is a double-stranded (ds)-DNA molecule. These conventional microarrays permit gene-expression investigation, which only tells a part of the genomic story.
TSCg, TSCp, TSPg, and TSPp and traditional chemogenomics, chemoproteomics, pharmacogenomics, and pharmacoproteomics2–6 show promise in biochemistry, molecular genetics, cell biology, discovery of novel gene–drug complexes, protein–drug target interactions, gene–protein–drug target sites, synthesis/sequencing of DNA and RNA, improving the characterization of transcription–transition, and enhancing drug discovery and development. TSCg, TSCp, TSPg, and TSPp can potentially lead to improvements in the benefit/harm balance of pathological therapies, and the identification of new pharmaceuticals.2–6
Chemogenomics, pharmacogenomics, chemoproteomics, and pharmacoproteomics
Conventional chemogenomics, pharmacogenomics, chemoproteomics, and pharmacoproteomics are all emerging approaches that will be used in the future to treat different pathologies.1–13 Chemogenomics is used in drug discovery to search for molecules that can interact with biological targets on a nucleic acid level. Chemoproteomics allows the effects of drug treatment and metabolism to be studied on proteins comprehensively. Pharmacogenomics is the part of pharmacology that deals with the influence of genetic variation on drug response in people by correlating the expression of genes or single-nucleotide polymorphisms with a drug’s efficacy or toxicological properties. Pharmacoproteomics is similar to pharmacogenomics, except that it involves the way pharmacology interacts with protein products.
Researchers know that drug response, toxicity, and efficacy fluctuate among people. The reason for this may be connected to pharmacokinetic factors (e.g., absorption, distribution, metabolism, and excretion). It may also be due to pharmacodynamic issues (e.g., interaction of the medicine on its target site).1–13
The objective of pharmacogenomics is to recognize patients who are most likely to be cured from a drug. Another goal is to isolate patients who are at elevated risk for unfavorable drug reactions. It is important for researchers to customize therapeutic interventions that maximize the benefit/risk relationship between patients and drug treatment. The ultimate goal of pharmacogenomics is to make personalized medicine, i.e., individualized medical treatment, possible.1–13
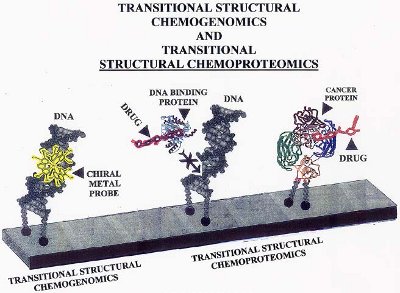
Figure 3 - Example of how TSCg and TSCp can be used with the novel DNA microarray to inhibit gene expression. The example with TSCg shows how a chiral metal probe binds to a specific region of a gene and inhibits gene (ds-DNA) expression by blocking ds-DNA-binding proteins. The examples with TSCp show how one DNA molecule is prevented from interacting with a DNA binding protein because a drug has attached to the active binding site of the protein. The second molecule shows how a cancer protein attached to DNA can be targeted by a drug under highly specific conditions.
Transitional structural chemogenomics and transitional structural pharmacogenomics
Genomics is the discipline of genetics concerned with the characterization of human and nonhuman genomes1–13 (i.e., DNA sequences) of organisms, mapping of genes, and functional parts of genes. TSCg and TSPg are new genomic methods that can be used with ultrasensitive small-molecule or large-molecule drugs and chemicals, respectively2–6 (Figure 3). Both techniques can be used to target traditional (e.g., right-handed ds-B-DNA), multistranded (triplex DNA), and alternative (cruciform DNA) nucleic acid molecules.
Additionally, they target specific gene sequences, such as single or multiple gene segments, which undergo significant transitions (right-handed ds-B-DNA to left-handed ds-Z-DNA) in the structures of DNA or RNA. These new genomic techniques can also be used to target multiple genetic factors, sequentially inhibiting or enhancing gene expression.1–13
The authors’ group prefers small-molecule drugs over large because of their better pharmacokinetic properties. Small ultrasensitive drugs are defined as chemical entities that can be prescribed in small concentrations and have an elevated affinity for specific DNA or RNA target sites. An example of a small ultrasensitive drug is the anticancer agent WP900, a Z-DNA-targeted chiral-selective enantiomer. Examples of large-molecule drugs are anti-Z-RNA, anti-Z-DNA, anti-triplex DNA, and anti-quadruplex DNA antibodies, and peptide drugs.1–13
The difference between TSCg and TSPg is that TSCg can provide a more complete picture of how biological molecules work together with cells. TSPg is a division of pharmaceutics concerned with the influence of genetic difference on drug response in specific people. TSPg is the whole-genome application of pharmacogenetics, which characterizes single genetic (i.e., individual genes) interactions with drugs.1–13
About 50% of the target sites in oncology are inside of the human cell. Consequently, small-molecule drugs permit researchers and clinicians to arrive at these distant target sites. Both TSCg and TSPg can be used to experimentally regulate the expression of normal or mutated human or nonhuman genes. An example of this is to turn off/on or variably control (like a dimmer switch) genetic factors. Exotic types of DNA and RNA molecules (e.g., hairpin RNA) and multistranded nucleic acids (e.g., triplex DNA and quadruplex DNA) can be directly targeted by employing TSCg and TSPg.1–13
The main purpose of TSCg and TSPg is to inhibit the expression of genetic elements by means of one or more drugs that target specific areas of genes (regulatory regions) that may experience structural changes. Examples of these regions are enhancers, basal promoters, and upstream promoters. Proteins and/or drugs that alter the genomic area of concern can be employed by TSCg and TSPg.
It should be noted that some genes contain very short segments of an alternate form of ds-DNA called left-handed Z-DNA. Genes that have the potential for B-DNA to Z-DNA transitions are most suitable for use with both TSCg and TSPg. Nevertheless, TSCg and TSPg can be used with genes that do not have Z-DNA sequences in them, namely, genes that only have conventional right-handed ds-B-DNA. From a mechanical perspective, the drug interacts with a specific ds-DNA or RNA sequence, directly blocking access to a gene by regulatory DNA- (or RNA-) binding proteins, or cutting (i.e., destroying) the DNA or RNA segment.1–13
TSCg and TSPg can be used by researchers to prevent the binding of other molecules to a range of different DNA and RNA molecules, such as DNA polymerase, RNA polymerase, topoisomerases, and nucleases. The structure and function of linked nucleic acids (LNA), antisense nucleic acids, and peptide nucleic acids (PNA)1–13 can be studied with TSCg and TSPg. LNA and PNA offer new opportunities to inhibit gene expression. In addition, antisense nucleic acid technology can be used to target many different types of DNA, RNA, and nucleic acid–protein complexes, such as triple-stranded DNA (i.e., forming a four-stranded DNA molecule) and quadruplex DNA (forming a five-stranded DNA molecule). The addition of another strand will directly down-regulate gene expression.1–13
The authors are working on several research projects involving, for example, skin cancer and cataracts, using TSCg and TSCp and the next generation of microarrays, viz., multistranded and alternative DNA, RNA, and plasmid microarrays (transitional structural nucleic acid microarrays).1–13 One study involves the augmentation of a gene. Up-regulation of genes can be very beneficial (e.g., a 10–20% increase in the usual levels of coagulation factor IX can alleviate the effects of hemophilia). The coagulation factor IX gene has Z-DNA sequences. Data reveal that a chemical and/or drug and/or protein targeted to this area (i.e., Z-DNA segment) of the coagulation factor IX gene controls gene expression.
The research team is also working with the normal bovine zeta-crystallin gene of the mammalian eye lens (i.e., different post-translational modifications of the protein). Results of the study show that TSCg and TSPg can be used with the bovine zeta-crystallin protein to target specific lefthanded Z-DNA segments in other genes. As a result of this interaction, bovine zeta-crystallin can be used to down-regulate expression of certain genetic factors.
TSCg and TSPg have also produced preliminary data on the characterization of some normal and mutated genes (e.g., Fragile X syndrome, FMRI gene [trinucleotide repeats]). The research team’s latest project involves four-stranded DNA (quadruplex DNA). Quadruplex DNA is found in the promoter region of some oncogenes. Targeting these oncogenes could prove to be an effective anticancer therapy. For example, the human c-Myc gene is an oncogene and its four-stranded DNA acts as a transcriptional regulator.1–13
Both TSCg and TSPg can also be applied to target and identify very small differences in DNA and RNA as they begin to undergo structural changes (e.g., right-handed A-RNA to left-handed Z-RNA). These differences in structure are characterized by biothermodynamics. Additionally, the authors were able to regulate gene expression by studying the specific conformational parameters (pitch heights, base tilt, and torsion angle) of nucleic acids that undergo transitions in structures.14–17
It is not enough to use chemicals that target specific base pairs of DNA, RNA, nucleic acid–protein complexes, and generalized proteins. Pharmacogenomics, pharmacoproteomics, chemogenomics, and chemoproteomics need to evolve to the next level—targeting explicit changes in nucleic acid and protein structure, and nonconventional nucleic acid molecules. Consequently, chemical/drug probes that target extremely specific changes in the configurations of the ss-DNA (ss-RNA) and ds-DNA (ds-RNA), alternative DNAs and RNAs, multistranded DNAs and RNAs, and proteins during different stages of structural modification (e.g., post-translational) need to be discovered and used. These probes/novel drugs must be able to specifically target gene expression in eukaryotic and prokaryotic cells and abnormal tissues.
TSCg and TSPg can also be used to study the aging process, that is, enhance genes that have undergone down-regulation due to DNA damage from aging (e.g., telomeres). Genomic factors can be best distinguished using high-throughput single-cell genomic characterizations. TSCg and TSPg allow for the examination of single-cell genomes via the use of novel nucleic acid-based microarrays (i.e., multistranded and alternative DNA, RNA, and plasmid microarrays [transitional structural nucleic acid microarrays]).1–13,18–20
Transitional structural chemoproteomics and transitional structural pharmacoproteomics
Proteomics is the large-scale examination of the structures and functions of protein molecules. 1–13 TSCp and TSPp (Figure 3) are proteomic methods that employ small-molecule or large-molecule chemicals and/or drugs, respectively, to target specific structures (e.g., active sites) of nucleic acid binding proteins, nontranscription factors such as kinases, or nonchromosomal proteins (molecular chaperones such as calnexin).1–3 These targeted sites include, but are not limited to, ss-nucleic acid, ds-nucleic acid, triplex-nucleic acid, and quadruplex-nucleic acid binding proteins.
TSCp and TSPp can be employed to study the discovery and development of biologicals: epitope mapping (protein–protein or protein–peptide interface mapping), structural biology (identifying the effect of point mutations on protein stability), and drug discovery (binding site and pharmacology correlations). TSCp and TSPp target proteins before, during, and after post-translational modifications. Both approaches target structured and/or unstructured regions of protein molecules.1–3
TSPp provides new ways to examine the influence of genetic variation on drug response in different patients,1–13 achieved by correlating gene expression (or single-nucleotide polymorphisms) with a drug’s efficacy or toxicity. TSPp works by chemically targeting a specific region of a transcription element (e.g., DNA-binding domain) at a certain point in time, which alters a protein’s structure and thereby changes its function. The targeted transcription factor can no longer recognize and bind to the DNA of interest, such as a promoter. The modified protein molecule can then be used to regulate the expression of a gene (e.g., mutated) by turning it off/on or variably controlling it. Variably controlling a gene has potential clinical value. TSCp involves protein–drug interactions within cells and tissues, and TSPp the characterization of genetic variation a drug has on a protein that regulates DNA or other proteins1–13 relative to its relationship with proteins.
TSCp and TSPp can target a cancer by changing the basic structure of the cancer protein, which in turn inhibits the pathology.1–13 The methods can also be used with a protein to deliver a drug or chemical that obliterates the cancer. In regard to nucleic acid–protein, nucleic acid–protein–protein, and nucleic acid–drug binding kinetics, it is important to scrutinize the gradual transition of one DNA or RNA structure to another nucleic acid conformation (e.g., B-DNA to Z-DNA).14–17
Science has come into an exhilarating period where proteomics are being used to study the mechanisms of drug interactions in patients. Additionally, proteomics is allowing for recognition of many different biomarkers for forecasting response to drug treatment. Consequently, proteomics will allow for the development of personalized medical treatments of patients with many different pathologies.
TSCp and TSPp will help us to better understand in vivo DNA–protein, in vivo RNA–protein, DNA–drug, RNA–drug, DNA–protein–drug, RNA–protein–drug, DNA–protein–protein interactions, RNA–protein–protein interactions, DNA–protein–protein–drug interactions, and RNA–protein–protein–drug interactions.1–12 TSCp and TSPp will assist in refining the discovery of proteomic interaction profiles of leads, hits, and drugs, and allow researchers to develop a more multifaceted image of the mechanisms of drug metabolism.
Exotic nucleic acids: Alternative and multistranded DNA and RNA structures
DNA and RNA are not static one-dimensional macromolecules. These nucleic acids are very dynamic entities with many exotic types of structures.14–17 Alternative and multistranded nucleic acids have specific functions in cells. These vibrant molecules undergo many structural changes and have the ability to bind with many different molecules and chemicals.
To appreciate how TSCg, TSPg, TSCp, TSPp (Figure 3), and the next generation of DNA and RNA microarrays (multistranded and alternative DNA, RNA, and plasmid microarrays [transitional structural nucleic acid microarrays]) (Figures 1 and 2) work is vital to understanding the structure and function of alternative and multistranded DNA and RNA structures. The majority of DNA in a cell is in the right-handed ds-B-DNA conformation. ds-Z-DNA is an alternative left-handed structure of ds-B-DNA. The potential B-DNA to Z-DNA transition exists transiently and is only found in some genes. Z-DNA is located in many human and nonhuman genes and performs different functions, such as in genomic rearrangements, as a transcriptional regulator, controlling gene expression, and RNA editing.14–17
Z-DNA is believed to be one of three different types of ds-DNA molecules with a specific biological function (the other two being A-DNA and B-DNA). Research has shown that the Z-DNA binding domain of ADAR1 may localize this particular enzyme that is used for modifications of the sequence of the newly synthesized RNA molecule to sites of active transcription zones. The expansion of ultrasensitive nucleic acid chemical probes for B-DNA and Z-DNA will become potent tools for regulating gene expression, since about 48% of human genes have DNA with the potential for a B-DNA to Z-DNA and Z-DNA to B-DNA helical transition.14–17
RNA also has the potential to take on exotic structures with specific biological functions. RNA has different configurations (e.g., messenger [m]-RNA, ribosomal [r]-RNA, and transfer [t]-RNA). This nucleic acid molecule can also change its structure and transition to other configurations. One such structure is right-handed A-RNA changing to left-handed Z-RNA.14–17 This molecule and its transitions therefore have great therapeutic potential.
Inhibitors to m-RNA, r-RNA, and t-RNA are even more valuable than drugs that directly inhibit DNA. Since RNA bases have more diverse pairings than DNA, they make perfect target sites for ultrasensitive probes. Furthermore, DNA molecules are situated in the cell’s nucleus, while RNA is located in the cytoplasm and nucleus. The cellular location of RNA is more accessible and therefore easier to target therapeutically.1–3
The targeting of multistranded nucleic acid molecules, such as quadruplex DNA (or RNA) and triplex DNA (or RNA) by specific ligand molecules, can also be involved in the drug development process. Additional ultrasensitive DNA and RNA probes need to be discovered that interact with triplex nucleic acids and quadruplex nucleic acids.1–3 One approach is to target telomerase with compounds that bind to the G-quadruplex DNA, such as porphyrins, perylene diimides, and diamidoanthraquinones.
TSCg, TSCp, TSPg, TSPp; traditional pharmacogenomics, pharmacoproteomics, chemogenomics, and chemoproteomics; and next-generation DNA microarrays (multistranded and alternative DNA, RNA, and plasmid microarrays [transitional structural nucleic acid microarrays]) will allow researchers to isolate new genomic and proteomic target sites based on structural transitions of DNA and RNA molecules, alternative and multistranded DNAs and RNAs, nucleic acid–protein complexes, and nucleic acid–protein–protein interactions.1–13,18–20 The end result of this union of different genomic and proteomic techniques will be the creation of novel target sites for new and established drugs used in the treatment of many different diseases. TSCg, TSCp, TSPg, and TSPp have the ability to characterize alternative and multistranded nucleic acids (and their respective interactions with nucleic acid binding proteins), namely, non-BDNA structures such as cruciform DNA, hairpin nucleic acids, triplex DNA, lefthanded Z-forms of DNA and RNA, and A-motif nucleic acids.16
Future directions in traditional pharmacogenomics, and TSCg, TSCp, TSPg, and TSPp
Pharmacogenomics is the study of how a human’s genetic inheritance affects a person’s response to drugs. It is still in its infancy and has many limitations. TSCg, TSCp, TSPg, and TSPp will enhance drug discovery and development and lower research and development costs.1–3 The most significant advancement will be the reduced risk of adverse drug reactions. Pharmacogenomics, along with bioinformatics, will find wider application by scientists and physicians during the early phases of clinical drug development. People with a variety of different pathologies and their physicians will benefit from pharmacogenomics because the drugs will be tailored for the most favorable health effects.
The combination of traditional pharmacogenomics and TSCg, TSCp, TSPg, and TSPp significantly impact the drug discovery and development process, advancing our knowledge of genetics, biochemistry, molecular biology, and pathology,1–3 and will help researchers better understand the effects of drugs on patients and the molecular biology of pathologies. In addition, it will help us more fully comprehend the effects of drug formulation on both efficiency and safety. This approach will make it possible to precisely optimize drug therapy based on an individual’s ability to metabolize, transport, and directly respond to prescribed drugs.1,2
Pharmacogenomics has limitations, and it must develop beyond the labeling of specific target sites as being either denatured ss-DNA, conventional ds-DNA, and specific base pairs that make up the majority of DNA (e.g., B-DNA). Additional limitations are the availability and cost of pharmacogenetic testing. TSCg, TSCp, TSPg, and TSPp will take pharmacogenomics from a one-dimensional to a multidimensional means of thinking about macromolecules, namely, DNA, RNA, protein, and nucleic acid–protein complexes.
TSCg, TSCp, TSPg, and TSPp, used together with the multistranded and alternative DNA, RNA, and plasmid microarrays (transitional structural nucleic acid microarrays), are the next phase in the growth of genomics and proteomics, and will permit the development of novel methods focused on improving nucleic acid-based research.1–3
Genomics and proteomics are evolving at an incredibly fast speed. Scientists, clinicians, and pharmaceutical researchers are generating genome- and proteome-wide results on increasingly larger study populations. Access to these results is increasing our knowledge of gene expression and human pathology. Translational research is converting data into diagnostic tools, target sites for drug development, and novel ways to treat and prevent human diseases.
Novel genomic and proteomic approaches will produce vibrant strategies that will optimize pharmacogenomics and transform the way scientists discover treatments and cures for such diseases as AIDS, cancer, depression, schizophrenia, Xeroderma pigmentosum, tuberculosis, asthma, viral infections, and genetic and cardiovascular diseases.1–3 At the same time they will enable a different approach for the characterization of differential gene expression and the transcriptome.
References
- Gagna, C.E.; Lambert, W.C. Novel multistranded, alternative, plasmid and helical transitional DNA and RNA microarrays: implications for therapeutics. Pharmacogenomics2009, 10(5), 895–914.
- Gagna, C.E.; Lambert, W.C. Novel drug discovery and molecular biological methods via DNA, RNA and protein changes using structure–function transitions: transitional structural chemogenomics, transitional structural chemoproteomics and novel multi-stranded nucleic acid microarray. Med. Hypotheses2006, 67, 1099–114.
- Gagna, C.E.; Lambert, W.C. Novel drug discovery and molecular biological methods via DNA, RNA and protein changes using structure function transitions: transitional structural chemogenomics, transitional structural chemoproteomics and novel multi-stranded nucleic acid microarray. Med. Hypotheses2006, 67(5), 1099–114.
- Gagna, C.E.; Chan, N.J. et al. Transitional structural chemogenomics. Microsc. Microanal.2006, 12(suppl. 2), 414–15.
- Gagna, C.E.; Chan, N.J. et al. Novel Drug Discovery Techniques and DNA Microarray: Transitional Structural Chemogenomics and Transitional Structural Chemoproteomics. In: Recent Research Developments in Biophysics; Transworld Research Network: Kerala, India, 2006; Vol. 5; pp 133–46.
- Gagna, C.E.; Chan, N.J. et al. Structural transitional chemogenomics and structural transitional chemoproteomics: two novel drug discovery approaches. FASEB J.2005, 19(4), A853; poster-521.4, Exp. Biol.2005.
- Gagna, C.E.; Lambert, W.C. Cell biology, chemogenomics and chemoproteomics—applications to drug discovery. Expert Opin.Drug Discov.2007, 2, 381–401.
- Gagna, C.E.; Winokur, D. et al. Cell biology, chemogenomics and chemoproteomics. Cell Biol. Inst.2004, 28, 755–64.
- Slaughter, R.L. Translation of pharmocogenetics to clinical practice: what will it take? Expert Rev. Clin. Pharmacol.2012,5(2), 101–3.
- Ong, F.S.; Deigan, J.L. et al. Clinical utility of pharmacogenetic biomarkers in cardiovascular therapeutics: a challenge for clinical implementation. Pharmacogenomics 2012, 13(4), 465–75.
- Degoma, E.M.; Rivera, G. et al. Personalized vascular medicine: individualizing drug therapy. Expert Opin.Drug Discov.2011, 16(5), 391–404.
- Roden, D.M.; Johnson, J.A. et al. Cardiovascular pharmacogenomics. Circ. Res.2011, Sept 16, 109(7), 807–20.
- Hudson, K.L. Genomics, health care, and society. N.Engl. J. Med. 2011, 365(11), 1033–41.
- Gagna, C.E.; Chan, N.J. et al. Localization and quantification of intact, undamaged right-handed double-stranded B-DNA, and denatured single-stranded DNA in normal human epidermis and its effects on apoptosis and terminal differentiation (denucleation). Archives Dermatological Res. 2009, 301, 659–72.
- Gagna, C.E.; Kuo, H.R. et al. Novel DNA staining method and processing technique for the quantification of undamaged double-stranded DNA in epidermal tissue sections by PicoGreen probe staining and microspectrophotometry. J. Histochem. Cytochem.2007, 55, 999–1014.
- Gagna, C.E.; Lambert, W.C. The halting arrival of left-handed Z-DNA. Med. Hypothesis2003, 60, 418–23.
- Gagna, C.E.; Winokur, D. et al. The Use of Histotechnology, Histochemical and Immunohistochemical Nucleic Acid Probes to Qualify and Quantify Cell Death in Single-, Double-, and Multiple-Stranded DNA in Diseased and Normal Tissues. In: Recent Research Developments in Biophysics; Transworld Research Network: Kerala, India, 2004; Vol. 3; pp 207–33.
- Method for Immobilizing Multistranded Nucleic Acid Molecules by Modifying More Than One Strand Thereof, and Binding Each Strand to a Solid Support. U.S. Patent 60/308,936, Aug 30, 2005.
- Method for Immobilizing Multistranded Nucleic Acid Molecules by Modifying More Than One Strand Thereof, and Binding Each Strand to a Solid Support. U.S. Patent 2003-517287, Oct 2, 2008.
- Gagna, C.E. Structural nucleic acid microarrays. Biochemistry2001, 40(29), 8648.
Dr. Gagna is an Associate Professor and Director of Biology Laboratories at New York Institute of Technology (NYIT), Department of Life Sciences, Theobald Hall, Room 430, 250 Valentines Ln., Old Westbury, NY 11568-8000, U.S.A., with his research lab at the New York College of Osteopathic Medicine (NYCOM) (Room 216B), Old Westbury, NY. He is also Adjunct Associate Professor, Department of Pathology & Laboratory Medicine, and Adjunct Assistant Professor, Department of Medicine (Dermatology), New Jersey Medical School, Newark, NJ, U.S.A.; tel.: 201-394-5304; fax: 516-686-3853; e-mail: [email protected]. Dr. W. Clark Lambert is a Professor in the Department of Pathology, and Department of Medicine (Dermatology), New Jersey Medical School, Newark, NJ. Mr. Peter C. Lambert is a medical student at St. George’s University School of Medicine, St. George’s, Grenada, West Indies. This work was supported in part by NYIT-ISRC grants (2010 and 2011) and instrumentation purchased from an MBIA Foundation Grant. The authors would like to thank the following: Mr. Greg Banhazl (Director of Business Development, NYIT) for his help in this project; Ms. Justine Chen, Ms. Sarah Syed, Mr. Mohammed Islam, and Ms. Christina Yohannan for their assistance in preparing the manuscript; and Future Medicine Ltd. (London, U.K.) for permission to reproduce Figures 1 and 4.